How to Get Rid of Black Lips From Smoking Weed
Introduction
This chapter summarizes the state of knowledge about the chemistry and toxicology of cigarette smoke and provides data relevant to the evaluations and conclusions presented in the disease-specific chapters of this report. The literature reviewed in this chapter is limited to manufactured cigarettes and does not include publications on handmade ("roll your own") cigarettes or other products that contain nicotine. The next section, "Chemistry," includes a brief description of technologies used by cigarette manufacturers in a limited number of cigarette brands marketed as "reduced-exposure" or "lower-yield" products. These commercial products have not been met with widespread consumer acceptance. The following section, "Biomarkers," focuses on the manufactured tobacco-burning cigarette consumed by the majority of smokers in the United States and elsewhere.
The section on "Chemistry" describes the chemical components of cigarette smoke and addresses aspects of product design that alter the components of cigarette smoke and factors affecting delivery of smoke to the smoker. In most cases, the data reported for chemical levels in mainstream smoke were derived under standard smoking conditions described by the U.S. Federal Trade Commission (FTC) and the International Organization for Standardization (ISO). These standard conditions are puff volume of 35 milliliters (mL), two-second puff duration, one-minute puff frequency, and butt length defined as either 23 millimeters (mm) for nonfilter cigarettes or the length of the filter overwrap paper plus 3 mm. When alternative smoking regimens are used, levels of potentially harmful substances in smoke emissions usually differ from those measured under standard conditions. (For more details, see "Delivery of Chemical Constituents into Tobacco Smoke" later in this chapter.) When people smoke, they do not use the puff volume and puff frequency programmed into smoking machines, and smoking habits vary significantly from person to person and cigarette to cigarette. Consequently, actual exposures to and doses of components of smoke cannot be derived from values obtained with machine smoking.
The section on "Biomarkers" offers an overview of in vitro and in vivo data on genotoxicity and cytotoxicity and a review of the literature on animal bioassays, in addition to general concepts of biomarkers of exposure, of biologically effective dose, and of potential harm, as an introduction to more detailed descriptions of biomarkers in subsequent chapters of this Surgeon General's report.
Cigarette smoke is a complex mixture of chemical compounds that are bound to aerosol particles or are free in the gas phase. Chemical compounds in tobacco can be distilled into smoke or can react to form other constituents that are then distilled to smoke. Researchers have estimated that cigarette smoke has 7,357 chemical compounds from many different classes (Rodgman and Perfetti 2009). In assessing the nature of tobacco smoke, scientists must consider chemical composition, concentrations of components, particle size, and particle charge (Dube and Green 1982). These characteristics vary with the cigarette design and the chemical nature of the product.
Fowles and Dybing (2003) suggested an approach to identify the chemical components in tobacco smoke with the greatest potential for toxic effects. They considered the risk for cancer, cardiovascular disease, and heart disease. Using this approach, these investigators found that 1,3-butadiene presented by far the most significant cancer risk; acrolein and acetaldehyde had the greatest potential to be respiratory irritants; and cyanide, arsenic, and the cresols were the primary sources of cardiovascular risk. Other chemical classes of concern include other metals, N-nitrosamines, and polycyclic aromatic hydrocarbons (PAHs). This evaluation, along with the Hoffmann list of biologically active chemicals (Hoffmann and Hoffmann 1998), was used to select the chemicals reviewed in this chapter. Other chemical components with potential for harm will be identified as analysis of tobacco smoke becomes more complete and cigarette design and additives change.
Chemistry
Phases of Tobacco Smoke
Smoke from a burning cigarette is a "concentrated aerosol of liquid particles suspended in an atmosphere consisting mainly of nitrogen, oxygen, carbon monoxide and carbon dioxide" (Guerin 1980, p. 201). Researchers have also described cigarette smoke as a "lightly charged, highly concentrated matrix of submicron particles contained in a gas with each particle being a multicompositional collection of compounds arising from distillation, pyrolysis, and combustion of tobacco" (Dube and Green 1982, p. 42). Tobacco smoke is a complex and dynamic chemical mixture. Researchers have analyzed whole smoke or used chemical and physical means to separately examine the gas and particulate portions of tobacco smoke. The gas phase is defined as the portion of smoke that passes through a glass fiber filter of specified physical parameters, and the particulate phase refers to all matter captured by the glass fiber filter (Pillsbury 1969). Standard methods for analysis of tobacco smoke separate the two phases by using Cambridge glass fiber filters designed to collect aerosol particles of 0.3 micrometers (μm) or larger with an efficiency not less than 99 percent (Pillsbury 1969). Although these separate phases are an artificial construct, they are useful for describing the results of analysis of the components of cigarette smoke typically obtained by machine smoking. When people smoke cigarettes, the continuum of physical characteristics in smoke does not include the differentiation into specific fractions. The diameter of cigarette smoke particles constantly changes, and as the particles coalesce after their formation, they grow in diameter. However, in diluted smoke, loss of a volatile chemical matrix or other components may cause particles to shrink and changes in the particle size may alter the relative amounts of certain chemicals in the gas and particle phases (Guerin 1980).
Smoke formation occurs when the cigarette is lit and a puff is taken or when the cigarette smolders between puffs. Mainstream smoke is released from the butt end of the burning cigarette during puffing, and sidestream smoke emanates from the burning cigarette coal when it smolders (Guerin 1980). The air in the immediate vicinity of an active smoker contains a mixture of sidestream smoke, exhaled mainstream smoke, and any smoke that passes through the porous paper surrounding the tobacco (Löfroth 1989). A greater quantity of sidestream smoke is generated when the amount of tobacco burned during smoldering increases relative to the amount burned during puffing (Johnson et al. 1973b; Perfetti et al. 1998). Thus, the way the cigarette is smoked (e.g., puff volume and time between puffs) can alter the relative levels of mainstream and sidestream smoke (Perfetti et al. 1998).
In addition, the ratio of the levels of chemical components in sidestream smoke to their levels in mainstream smoke can be altered by differences among cigarettes (Perfetti et al. 1998). These differences are related to the tobacco blend or type, the tobacco preparation (e.g., cut width, additives, and moisture level), the dimensions of the cigarette, the weight of the tobacco rod, the porosity of the paper, the presence of a filter, and the type of filter. Studies using a machine that simulates human smoking have determined that the change in the ratio of sidestream to mainstream smoke components after introducing a filter and ventilation primarily resulted from a decrease in the amount of mainstream smoke, because the amount of sidestream smoke does not change substantially with alterations in cigarette design (Perfetti et al. 1998). Examination of chemicals with similar properties revealed that those with a low boiling point had higher ratios of levels in sidestream smoke to levels in mainstream smoke and that compounds with a high boiling point had lower ratios (Sakuma et al. 1984). Studies indicate that compared with mainstream smoke collected under standard FTC/ ISO smoking parameters, sidestream smoke has higher levels of PAHs (Grimmer et al. 1987; Evans et al. 1993); nitrosamines (Brunnemann et al. 1977a, 1980; Hoffmann et al. 1979a; Rühl et al. 1980); aza-arenes (Dong et al. 1978; Grimmer et al. 1987); aromatic amines (Patrianakos and Hoffmann 1979); carbon monoxide (CO) (Hoffmann et al. 1979b; Rickert et al. 1984); nicotine (Rickert et al. 1984; Pakhale et al. 1997); ammonia (Brunnemann and Hoffmann 1975); pyridine (Johnson et al. 1973b; Brunnemann et al. 1978; Sakuma et al. 1984); and the gas phase components 1,3-butadiene, acrolein, isoprene, benzene, and toluene (Brunnemann et al. 1990). With increased puffing intensity, the toxicant ratios of sidestream to mainstream smoke decrease (Borgerding et al. 2000).
The increase in the amount of tobacco burned during smoldering compared with tobacco burned during puffing is not the only factor influencing differences in the chemical content of sidestream and mainstream smoke. The burning conditions that generate sidestream and mainstream smoke also differ (Guerin 1987). Temperatures reach 900°C during a puff and fall to about 400°C between puffs (Guerin 1987). Puffing burns the tobacco on the periphery of the cigarette, and tobacco in the core burns between puffs (Johnson 1977; Hoffmann et al. 1979a). Thus, mainstream smoke depends on the chemical composition of the combustible portion of the cigarette near the periphery of the rod, whereas chemicals at higher concentrations in the central portion of the rod have higher levels in sidestream smoke than in mainstream smoke (Johnson 1977). Sidestream smoke is produced during conditions with less available oxygen (Guerin et al. 1987) and higher alkalinity and water content than those for mainstream smoke (Brunnemann and Hoffmann 1974; Adams et al. 1987; Guerin 1987). Ammonia levels are significantly higher in sidestream smoke, resulting in a more alkaline pH (Adams et al. 1987). Thus, the composition and levels of chemical species in mainstream smoke differ from those in sidestream smoke.
Levels of some compounds are higher in mainstream smoke than in sidestream smoke, and this difference may reflect chemical influences that are more complex than just changes in puff frequency. For example, mainstream smoke contains considerably more cyanide than side-stream smoke does (Johnson et al. 1973b; Brunnemann et al. 1977a; Norman et al. 1983). Sakuma and colleagues (1983) measured a series of semivolatile compounds in tobacco smoke and found that levels of phenol, cresol, xylenols, guiacol, formic acid, and acetic acid were higher in sidestream smoke, whereas levels of catechol and hydroquinone were higher in mainstream smoke.
Individual chemical constituents may be found in the particulate phase, the gas phase, or both (Guerin 1980). As cigarette smoke dissipates, chemicals may pass between the particulate and gas phases (Löfroth 1989). The gas phase contains gases and chemical constituents that are sufficiently volatile to remain in the gas phase long enough to pass through the Cambridge glass fiber filter (Guerin 1980), but as the filter becomes wet during the first puffs, hydrophilic compounds tend to adhere to it. The gas phase of cigarette smoke includes nitrogen (N2), oxygen (O2), carbon dioxide (CO2), CO, acetaldehyde, methane, hydrogen cyanide (HCN), nitric acid, acetone, acrolein, ammonia, methanol, hydrogen sulfide (H2S), hydrocarbons, gas phase nitrosamines, and carbonyl compounds (Borgerding and Klus 2005; Rodgman and Perfetti 2009). Constituents in the particulate phase include carboxylic acids, phenols, water, humectants, nicotine, terpenoids, paraffin waxes, tobacco-specific nitrosamines (TSNAs), PAHs, and catechols. Mainstream smoke contains only a small amount of nicotine in the gas phase (Johnson et al. 1973b; Pakhale et al. 1997), but the fraction of nicotine in the gas phase is higher in side-stream smoke because of the higher pH (Johnson et al. 1973b; Brunnemann and Hoffmann 1974; Adams et al. 1987; Pakhale et al. 1997). Brunnemann and colleagues (1977b) studied both mainstream and sidestream smoke and found that the gas phase of mainstream smoke contained more cyanide than did the particulate phase. Johnson and colleagues (1973b), however, showed that in sidestream smoke, cyanide is present almost exclusively in the particulate phase. Guerin (1980) concluded that both formaldehyde and cyanide may be present in both phases, and Spincer and Chard (1971) found formaldehyde in both the particulate and gas phases. The PAHs in the gas phase were only 1 percent of total PAHs, and the PAH distribution between gas and particulate phases varied with the boiling point of the PAHs (Grimmer et al. 1987). Because physical and chemical changes occur after tobacco smoke is drawn from the cigarette, some of the reported differences in PAH levels could result from differences in measurement techniques.
In summary, cigarette smoke is a complex and dynamic system. The concentration of smoke and the time after it leaves the cigarette can cause changes in particle size that may alter the relative amounts of certain chemicals in the gas and particle phases. Also, specific properties of the tobacco, the physical design of the cigarette, and the machine-smoking method that is employed to generate mainstream smoke for analyes can have a significant impact on the levels of both mainstream and side-stream emissions.
Nicotine and Free Nicotine
The tobacco leaf contains many alkaloid chemicals; nicotine is the most abundant. Nicotine content varies, among other factors, by the leaf position on the tobacco stalk and also by the blend or leaf type used in a given cigarette or cigar (Tso 1990; Kozlowski et al. 2001). Plants such as tobacco that are characterized by high alkaloid content often possess a natural pharmacologic defense against microorganisms, insects, and vertebrates. For example, nicotine is toxic to many insects and, for many years, has been extracted from tobacco for use as a commercial pesticide (Domino 1999). Nicotine is addictive in humans because a portion of the nicotine molecule is similar to acetylcholine, an important brain neurotransmitter (Brody et al. 2006).
The alkaloids in tobacco leaf include anatabine, anabasine, nornicotine, N-methylanabasine, anabaseine, nicotine, nicotine N′-oxide, myosmine, β-nicotyrine, cotinine, and 2,3′-bipyridyl (Figure 3.1). In commercial tobacco products, nicotine concentrations range from 6 to 18 milligrams per gram (mg/g) (0.6 to 1.8 percent by weight) (International Agency for Research on Cancer [IARC] 2004; Counts et al. 2005). Together, the sum of the concentrations of anatabine, anabasine, and nor-nicotine equals approximately 5 percent of the nicotine concentration (Jacob et al. 1999). Many minor tobacco alkaloids are pharmacologically active in humans in one or more ways. Clark and colleagues (1965) observed that some of these alkaloids had physiological effects in a variety of animal tests. Lefevre (1989) reviewed the evidence and concluded that anabasine and nornicotine had demonstrated effects on smooth muscle fiber, blood pressure, and enzyme inhibition. The literature on potentially addictive properties of these minor alkaloids is limited. S(-)-nicotine, which is present in the tobacco leaf, is structurally similar to forms of several minor alkaloids also found in the tobacco leaf, such as S(-)-N-methylanabasine (Figure 3.2). Moreover, Dwoskin and colleagues (1995) reported that in the rat, anatabine, anabasine, N-methylanabasine, anabaseine, and nornicotine all release dopamine from striatal brain tissue. Overall, it is likely that some of the minor tobacco alkaloids could (1) be addictive if delivered alone at sufficiently high levels and (2) act together with nicotine during tobacco use to generate effects that are difficult to discern because nicotine levels are so much higher. In addition to addictiveness, both nicotine and minor secondary amine alkaloids are precursors of carcinogenic TSNAs (IARC 2004, 2007).
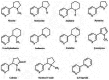

Figure 3.2
Structures of nicotine and minor alkaloid S(-)-N-methylanabasine in tobacco leaf.
The unprotonated nicotine molecule contains two nitrogen atoms with basic properties. The unprotonated nicotine molecule can thus add one proton to form a monoprotonated species or two protons to form the diprotonated species (Figure 3.3) (Brunnemann and Hoffmann 1974). The first proton added to nicotine attaches predominantly to the nitrogen on the five-membered (pyrrolidine) ring, because that nitrogen is significantly more basic than the nitrogen on the six-membered (pyridine) ring. Although protonated nicotine is not volatile, unprotonated nicotine is volatile and is able to enter the gas phase and readily pass into lipid membranes. Unprotonated nicotine is therefore free of the limitations that come with carrying an ionic charge, and the scientific literature and tobacco industry documents frequently refer to nicotine in this form as both "free nicotine" and "free-base nicotine." In the tobacco plant and in the dried leaf, nicotine largely exists in its ionic forms; otherwise, it would be rapidly lost to the surrounding atmosphere.

In water or in the droplets of particulate matter in tobacco smoke, the distribution of nicotine among its three forms depends on the pH of the solution. Increasing acidity of the solution increases the fraction of protonated molecules; conversely, increasing basicity increases the fraction in the unprotonated (free base) form (Figure 3.3). Because all forms of nicotine are highly soluble in water, all of the nicotine entering the respiratory tract from one puff of tobacco smoke easily dissolves in lung fluids and blood. However, because unprotonated nicotine from tobacco smoke particles is volatile, whereas protonated nicotine is not, a higher percentage of unprotonated nicotine in a puff results in a higher rate of nicotine deposition in the respiratory tract (Pankow 2001; Henningfield et al. 2004). The exact nature and effects of the increased rate of deposition depends on the chemical composition and the size of particles in the tobacco smoke, as well as topographic characteristics of smoking, such as puff size and duration and depth of inhalation. Increased rates of deposition in the respiratory tract lead to increased rates of nicotine delivery to the brain, which intensify the addictive properties of a drug (Henningfield et al. 2004). The conventional view has been that a sample of particulate matter from tobacco smoke is not usually so acidic that the diprotonated form becomes important. In water at room temperature, the approximate dividing line between dominance by protonated forms or by the unprotonated form is a pH of 8 (González et al. 1980). At higher pH, the fraction of unprotonated nicotine (αfb) is greater than the fraction of protonated nicotine (Pankow 2001). At pH 8, the two fractions are present in equal percentages. At any lower pH, the fraction of protonated nicotine is greater.
Because a typical sample of particulate matter from tobacco smoke collected from a cigarette or cigar is mostly nonaqueous liquid, it is not possible to take conventional pH measurements to determine nicotine distribution between the monoprotonated and unprotonated forms (Pankow 2001). However, it is possible to measure the concentration of unprotonated nicotine in a sample of tobacco smoke particulate (c p,u), because that level produces a directly proportional concentration of unprotonated nicotine in the gas phase, which is measurable (Pankow et al. 1997, 2003; Watson et al. 2004). Measuring the concentration of nicotine in a sample of tobacco smoke in the particulate phase (c p,t) allows calculation of the fraction of unprotonated nicotine: αfb = c p,u/c p,t (Pankow et al. 2003). To simplify the discussion of αfb values in tobacco smoke, Pankow (2001) introduced the term "effective pH" (pHeff), which refers to the pH needed in water to obtain the αfb value in a sample of particulate matter from smoke. Reported values of αfb for smoke from commercial cigarettes at 20oC were 0.006 to 0.36 (Pankow et al. 2003; Watson et al. 2004), which corresponds to pHeff values at 20oC in the range of 5.8 to 7.8.
The fraction αfb for particulate matter in tobacco smoke is important because the rapidity with which inhaled nicotine from tobacco smoke evaporates from the particulate phase and deposits on the linings of the respiratory tract is directly proportional to the αfb value for the smoke (Pankow et al. 2003). According to numerous tobacco industry documents, increasing levels of unprotonated nicotine in tobacco smoke was known to increase smoke "strength," "impact," "kick," and/or "harshness" (Backhurst 1965; Dunn 1973; Teague 1974; Ingebrethsen and Lyman 1991). Because of similar mechanisms, nicotine replacement therapy delivering gaseous nicotine caused throat irritation at delivery levels per puff that were similar to those reached by smoking a cigarette rated by using the FTC regimen at approximately 1 mg of total nicotine delivery; thus, cigarette design is focused on a balance between smoke "impact" and irritation. Some researchers have suggested that the irritation and harshness of smoke at higher pH makes it harder for smokers to inhale this smoke into the lungs (Brunnemann and Hoffmann 1974).
The value of αfb for particulate matter in each puff of smoke from one brand of cigarette or cigar strongly depends on the overall proportion of acids to bases in the puff (Pankow et al. 1997). As already noted, nicotine itself is a base. The natural acids in tobacco smoke (e.g., formic acid, acetic acid, and propionic acid) can protonate nicotine and tend to reduce αfb from its maximum of 1.0. The natural bases (e.g., ammonia) tend to neutralize the acids and keep more nicotine in the unprotonated form.
Variability in the acid-base nature of commercially available tobacco leaf is considerable. Flue-cured ("bright") tobacco is typically viewed as producing acidic smoke. Air-cured ("burley") tobacco is typically viewed as producing basic smoke. Simple adjustment of the tobacco blend can therefore produce a considerable range of acid or base content in tobacco smoke. In acidic smoke, αfb can be 0.01 or lower (e.g., 1-percent unprotonated nicotine), and in basic smoke, the αfb can be relatively high (e.g., 0.36 [36-percent unprotonated nicotine]) (Pankow et al. 2003; Watson et al. 2004).
Tobacco additives that are bases increase αfb values in mainstream smoke, and these additives are discussed extensively in tobacco industry documents (Henningfield et al. 2004). The documents reveal that a variety of basic additives have been considered, including ammonia and ammonia precursors. Conversely, some manufacturers also were interested in reducing harshness to a minimum and investigated acidic additives such as levulinic acid as "smoothing" agents. In that context, the natural basicity of a specific blend and the harshness of the smoke can be reduced by acidic additives such as levulinic acid, which tend to reduce αfb (Guess 1980; Stewart and Lawrence 1988).
In summary, nicotine in cigarette smoke exists in either a protonated or unprotonated form, depending on a number of factors, including the presence of natural acids and bases, the tobacco blend, tip ventilation, and the use of additives. Cigarette design ensures that the smoke has enough unprotonated nicotine to rapidly transfer nicotine into the body but not so much of it as to be too harsh for the smoker to continue to smoke.
N-Nitrosamines
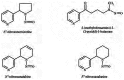
N-nitrosamines are a class of chemical compounds containing a nitroso group attached to an amine nitrogen. There are two types of nitrosamines in tobacco and tobacco smoke: volatile and nonvolatile, including TSNAs (Hoffmann et al. 1981; Tricker et al. 1991; Spiegelhalder and Bartsch 1996; IARC 2007). The volatile nitrosamines include N-nitrosodimethylamine, N-nitrosoethylmethylamine, N-nitrosodiethylamine, N-nitro-sopyrrolidine, and N-nitrosomorpholine. The nonvolatile nitrosamines are 4-(N-nitroso-N-methyl-amino)butyric acid, N-nitrosopipecolic acid, N-nitroso-sarcosine, 3-(N-nitroso-N-methylamino)propionic acid, N-nitrosoproline, and N-nitrosodiethanolamine. The nonvolatile TSNAs (Figure 3.4) have been examined extensively in tobacco and tobacco smoke. They include N′-nitrosonornicotine (NNN), 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), N′-nitrosoanatabine (NATB), and N′-nitrosoanabasine (NAB). The levels of nitrosamines in tobacco products are higher than are those in other consumer products, such as cooked bacon and beer (Hecht and Hoffmann 1988), and smokers are exposed to higher levels of TSNAs than of the other nitrosamines (Hoffmann et al. 1981; IARC 2007).
Studies have been conducted to identify precursors of nitrosamines and to determine the conditions required for their formation in tobacco. The primary intent of this research was to identify ways to reduce nitrosamine levels in tobacco and tobacco smoke. Secondary and tertiary amines in tobacco, including the alkaloids, react with nitrosating agents to form N-nitrosamines (Hecht and Hoffmann 1988). Hecht and colleagues (1978) showed that both nicotine and nornicotine can react with sodium nitrite under controlled conditions to form carcinogenic NNN and NNK, but nicotine is considered more important because of its higher level in tobacco products. TSNAs are not present at trace levels in freshly harvested tobacco, but they are predominantly formed during processing, curing, and storage (Hoffmann et al. 1974, 1981; Chamberlain et al. 1984; Andersen and Kemp 1985; Bhide et al. 1987; Djordjevic et al. 1989; Fischer et al. 1989b; Fisher 2000a). Aerobic bacteria play a major role in TSNA formation in air-cured tobacco (Hecht et al. 1975; Hoffmann et al. 1981; Parsons et al. 1986). In flue-cured tobacco, the curing conditions alter levels of nitrosamines (Fisher 2000a). Before the late 1960s and early 1970s, direct-fire curing in the United States did not produce high levels of TSNAs. When propane gas was introduced as the combustion source (Fisher 2000a), nitrogen oxides from the exhaust gases in tobacco barns reacted with alkaloids in the tobacco plant to form TSNAs. Hoffmann and colleagues (Hoffmann et al. 1981; Brunnemann and Hoffmann 1991) also revealed that N-nitrosodiethanolamine is formed from the diethanolamine used in the formulation of maleic hydrazide, which is applied to regulate suckers on tobacco plants.
Volatile nitrosamines are found primarily in the gas phase of tobacco smoke, and TSNAs are almost exclusively found in the particulate phase (Guerin 1980). Researchers suggest that about one-half of the nitrosamines in tobacco smoke are transferred unchanged from the tobacco to the smoke and that the remainder is formed from pyrosynthesis during smoking (Hoffmann et al. 1977; Adams et al. 1983). Other researchers have concluded that almost all TSNAs are transferred directly from the tobacco (Fischer et al. 1990b).
It is difficult to determine whether TSNAs are pyrosynthesized or transferred intact, because the most important factors in nitrosamine formation such as concentrations of preformed TSNA in tobacco or their precursor, as well as chemical and physical processes during smoking, could affect either mechanism. Morie and Sloan (1973) reported that the nitrate and amine content in tobacco determined the amount of N-nitrosodimethyl-amine formed in tobacco smoke. This finding has been widely duplicated by researchers looking at other nitrosamines (Hecht et al. 1975; Brunnemann et al. 1977a, 1983; Hoffmann et al. 1981; Adams et al. 1983, 1984; Fischer et al. 1989b; Tricker et al. 1991; Atawodi et al. 1995; Spiegelhalder and Bartsch 1996). Other factors that influence nitrate concentrations in tobacco can also indirectly influence nitrosamine concentrations. Because TSNA content is strongly influenced by the use of stems that are naturally high in TSNAs in the cigarette rod, the increased use of stems leads to higher nitrosamines in the smoke (Brunnemann et al. 1983). Researchers have also found that the use of nitrogen fertilizer can contribute to the concentration of nitrosamines in tobacco and ultimately in the smoke (Johnson and Rhoades 1972; Tso et al. 1975; Brunnemann et al. 1977a; Chamberlain et al. 1984, 1986). Other influential factors identified were tobacco growth conditions, storage times, storage temperatures (Andersen et al. 1982; Andersen and Kemp 1985), and the stalk positions from which the tobacco leaves are harvested (Chamberlain et al. 1986).
Another factor contributing to nitrosamine concentrations in tobacco is the type of tobacco used (Johnson and Rhoades 1972; Brunnemann et al. 1983; Fischer et al. 1989b,c). Oriental tobaccos are lowest in both nitrates and TSNAs (Fischer et al. 1989b), whereas burley tobacco contains the highest TSNA concentrations (Fischer et al. 1989b,c). The nitrosamine concentrations in bright tobacco are between those in oriental and burley and depend on the curing practices described earlier (Tso et al. 1975; Hoffmann et al. 1979a). The TSNA concentrations are higher in blended cigarettes than in those made from bright tobacco, because burley is included in the blend (Fischer et al. 1990a). In most tobaccos, NNN concentrations exceed NNK concentrations (Fischer et al. 1989b), but in bright tobacco, NNK concentrations exceed those of NNN (Fischer et al. 1989b, 1990a).
The preformed concentration of nitrosamines in tobacco leaves and stems is a major determinant of the levels in tobacco smoke (Fischer et al. 1990c; Spiegelhalder and Bartsch 1996). However, for cigarettes that have the same concentrations of nitrosamines in the tobacco, the nitrosamine levels in the smoke were largely determined by the degree of ventilation and the use of cellulose-acetate filter tips in the cigarette. After examining machine-generated smoke, by the FTC/ISO method, from cigarettes containing the same type of tobacco, whether blended or bright only, researchers found that nitrosamine levels are correlated with tar delivery, which is primarily a function of filter ventilation (Adams et al. 1987; Fischer et al. 1990a). However, studies of cigarettes with different blends of tobacco have shown that tar is not an accurate measure of nitrosamine levels (Fischer et al. 1989c; Spiegelhalder and Bartsch 1996; Counts et al. 2004). Studies have also shown that cellulose-acetate filter tips remove both volatile nitrosamines and TSNAs (Morie and Sloan 1973; Brunnemann et al. 1980; Rühl et al. 1980; Hoffmann et al. 1981). These findings indicate the importance of measuring TSNA levels in smoke, rather than using measured levels of tar or nicotine to predict levels of TSNAs in smoke on the basis of an average relationship between tar or nicotine and TSNAs.
Nitrosamine levels measured in the tobacco and the smoke from cigarettes that were purchased around the world vary widely because of the differences cited above. Historically, the ranges of levels of NNN (2 to 12,454 nanograms [ng] per cigarette), NAB+NATB (109 to 1,033 ng), and NNK (55 to 10,745 ng) in cigarette tobacco were wide (Hoffmann et al. 1974; Fischer et al. 1989b, 1990a,c; Tricker et al. 1991; Atawodi et al. 1995; IARC 2004, 2007). More recent analyses have given more consistent results that depend on the blend of tobacco (NNN + NNK: 87 to 1,900 ng/g) (Ashley et al. 2003). Levels in mainstream tobacco smoke, as reported by the FTC/ISO machine-smoking method, have been reported at an order of magnitude lower than those in tobacco (NNN = 4 to 1,353 ng generated per cigarette); NAB+NATB = 10 to 82 ng; and NNK = 5 to 1,749 ng (Fischer et al. 1989b, 1990a,c; Tricker et al. 1991; Atawodi et al. 1995; Mitacek et al. 1999).
Using the ISO, Massachusetts (MDPH; 45-mL puff volume, 30-second puff interval, 50 percent of ventilation holes blocked) and Canadian Intense (CAN; 55-mL puff volume, 30-second puff interval, 100 percent of ventilation holes blocked) smoking regimens, Counts and colleagues (2005) reported the levels of TSNAs in mainstream smoke from Philip Morris cigarettes sold internationally. The investigators found that in mainstream smoke, NNN levels were 5.0 to 195.3 ng generated per cigarette for ISO, 16.3 to 374.2 ng for MDPH, and 20.6 to 410.6 ng for CAN. NNK levels were 12.4 to 107.8 ng generated per cigarette for ISO, 25.8 to 206.6 ng for MDPH, and 39.1 to 263.0 ng for CAN. NATB levels were 8.0 to 160.4 ng generated per cigarette for ISO, 31.9 to 295.3 ng for MDPH, and 43.5 to 345.1 ng for CAN.
The combined levels of NNN and NNK reported by Wu and associates (2005) are in good agreement with the ranges reported by Counts and colleagues (2005). This finding suggests that the more advanced analytical methods used in these later studies yielded more accurate measures for current cigarettes than did previous measures. Levels of volatile nitrosamines in mainstream tobacco smoke are typically lower than those of the TSNAs (dimethylnitrosamine = 0.1 to 97 ng generated per cigarette; methylethylnitrosamine = 0.1 to 9.1 ng; and N-nitrosopyrrolidine = 1.5 to 64.5 ng) (Brunnemann et al. 1977a, 1980; Adams et al. 1987).
Ashley and colleagues (2003) compared TSNA concentrations in tobacco from Marlboro cigarettes with those in locally popular, non-U.S. brands of cigarettes in 13 countries. For most of the countries, TSNA concentrations in the tobacco from Marlboro cigarettes were higher than those in tobacco from locally popular brands from that country. TSNA concentrations varied widely (20-fold overall) between and within brands from the same country and differed significantly from country to country. This study confirmed earlier work showing wide variations in TSNA levels in tobacco and smoke from products within a country and between countries (Hecht et al. 1975; Fischer et al. 1990c; Spiegelhalder and Bartsch 1996; Gray et al. 2000). The basic findings from this study were confirmed by work from Wu and colleagues (2005), who examined combined levels of NNN and NNK in the mainstream smoke from cigarettes from the same 13 countries and also found a wide variation in this matrix.
Identification of growing, curing, and blending practices that alter nitrosamine levels in tobacco and smoke have led researchers to agree that low TSNA levels in smoke can be achieved by using particular varieties of tobacco and carefully controlling the factors leading to formation and transfer of TSNAs from tobacco into smoke (Brunnemann et al. 1977a; Hoffmann et al. 1977; Hecht et al. 1978; Rühl et al. 1980; Andersen and Kemp 1985; Hecht and Hoffmann 1988; Fischer et al. 1990c; Spiegelhalder and Bartsch 1996; Mitacek et al. 1999; Ashley et al. 2003; Burns et al. 2008). To reduce TSNAs, tobacco curing in the United States is undergoing a transition, and nitrosamine levels may change as curing and blending practices change (Counts et al. 2004; O'Connor et al. 2008).
In summary, nitrosamines are found in tobacco and tobacco smoke at high levels compared with other consumer products. The levels of these compounds, which are formed during tobacco processing, curing, and storage, can be minimized by breeding and selecting tobacco lines with lower propensity for TSNA formation, and limiting the use of nitrogen fertilizer, the levels of nitrogen oxides in the atmosphere during curing, the amount of burley tobacco in the blend, and storage times. The impact of different practices is clearly seen by the wide global range of TSNA levels in tobacco and smoke.
Polycyclic Aromatic Hydrocarbons
PAHs are chemical compounds with two or more condensed aromatic and other cyclic rings of carbon and hydrogen atoms (Douben 2003). Recent studies (Rodgman and Perfetti 2006) have identified at least 539 PAHs in tobacco smoke. The U.S. Environmental Protection Agency (EPA) has identified 16 priority environmental PAHs on the basis of evidence that they cause or may cause cancer: acenaphthylene, acenaphthene, anthracene, benz[a] anthracene, benzo[a]pyrene (B[a]P), benzo[b]fluoranthene (B[b]F), benzo[k]fluoranthene (B[k]F), benzo[g,h,i] perylene, chrysene, dibenz[a,h]anthracene, fluoranthene, fluorene, indeno[1,2,3-cd]pyrene, naphthalene, phenanthrene, and pyrene (Figure 3.5) (USEPA 1980, 1986). The 16 PAHs, which have two to six fused rings and molecular weights of 128 to 278, were detected in the particulate matter of tobacco smoke (IARC 1986, 2004; Ding et al. 2006, 2007). PAHs range from highly volatile to relatively nonvolatile, and their distribution in the particulate and gas phases of tobacco smoke varies with the boiling point (Grimmer et al. 1987). However, the gas phase contained only an estimated 1 percent of the total PAHs found in tobacco smoke. The composition of PAHs in mainstream smoke is different from that in sidestream smoke (Grimmer et al. 1987), and the lipophilic characteristics range from moderate to high (Douben 2003).

Figure 3.5
Priority environmental polycyclic aromatic hydrocarbons.
PAHs are formed by incomplete combustion of natural organic matter such as wood, petroleum, and tobacco and are found throughout the environment (Evans et al. 1993; Douben 2003). In the burning cone at the tip of the tobacco rod, various pyrolysis reactions occur to form methylidyne (CH) radicals that are precursors to the pyrosynthesis of PAHs. Hoffmann and Wynder (1967) were the first to show that adding nitrate to tobacco reduced B[a]P levels. During smoking, nitrates form O2 and nitric oxide (NO), which intercept radicals and reduce PAH levels (Johnson et al. 1973a; Hoffmann and Hoffmann 1997). Other researchers also reported that the presence of nitrate in tobacco decreases B[a]P levels in the smoke (Torikai et al. 2005). The pyrolytic conditions also favor the formation of PAHs from certain isoprenoids such as solanesol (IARC 1986), although other findings have disagreed with this assessment (Torikai et al. 2005). B[a]P is the most widely known and studied PAH (IARC 2004).
Differences in tobacco type can affect levels of PAHs in the smoke. Flue-cured (bright) or sun-cured (oriental) tobaccos have lower nitrate content than does air-cured (burley) tobacco. Pyrosynthesis of PAHs generates higher PAH levels in smoke from cigarettes made exclusively with flue-cured or sun-cured tobaccos than in smoke from cigarettes made with burley tobaccos (Hoffmann and Hoffmann 1997; Ding et al. 2005). Cigarettes made from reconstituted tobacco with cellulose fiber as an additive yield significantly reduced PAH levels. Evans and colleagues (1993) measured PAHs in mainstream and sidestream smoke and found that B[a]P, B[b]F, and B[k]F levels are related to tar yields in cigarette smoke that result from differences in cigarette ventilation.
Some studies reported the levels of B[a]P alone as a surrogate for the total PAH content. Ding and colleagues (2005) observed that total PAH levels in mainstream smoke from commercial cigarette brands varied from 1 to 1.6 μg generated per cigarette under FTC machine-smoking conditions. In the same study, individual PAHs ranged from less than 10 ng generated per cigarette (B[k]F) to approximately 500 ng (naphthalene) (Ding et al. 2005). Other researchers reported levels of B[b]F at 10.4 ng, B[k]F at 5.1 ng, and B[a]P at 13.4 ng generated per cigarette (Evans et al. 1993). In four of five brands tested, B[a]P concentrations in cigarette tar were about 0.5 ng/ mg of tar (Tomkins et al. 1985). Kaiserman and Rickert (1992) reported the levels of B[a]P in smoke from 35 brands of Canadian cigarettes by using the ISO method; mean levels were 3.36 to 28.39 ng generated per cigarette. Although B[a]P levels were linearly related to declared tar values, the tar values and the B[a]P levels did not change at the same relative rate. In a study of PAHs in mainstream smoke from cigarettes from 14 countries, Ding and colleagues (2006) showed a significant global variation in levels. They also demonstrated an inverse relationship with TSNA levels at high PAH and low TSNA levels, possibly as a result of differences in nitrate levels.
In summary, PAHs result from the burning of biologic material, so they are present in the smoke from any form of burning tobacco. Factors that can affect PAH levels in tobacco smoke include the type of tobacco and its nitrate content. Because of divergent pyrosynthetic mechanisms, factors that increase the nitrate content of tobacco decrease PAH levels but may increase TSNA levels in cigarette smoke. However, a substantial reduction in PAH levels in cigarette smoke will be a challenge as long as tobacco smoke is generated from burning tobacco.
Volatile Compounds Including Aldehydes
When a cigarette is smoked, chemicals partition between the particulate and gas phases on the basis of physical properties including volatility and solubility (Hoffmann and Hoffmann 1997). Complete partitioning of any chemical to the gas phase of cigarette smoke is generally limited to the gaseous products of combustion, such as the oxides of nitrogen, carbon, and sulfur, and the extremely volatile low-molecular-weight organic compounds. There are between 400 and 500 volatile gases and other compounds in the gas phase (Hoffmann and Hoffmann 1997). The nearly complete combustion of the cigarette tobacco filler generates an effluent stream of gaseous chemicals residing almost exclusively in the gas phase portion of mainstream cigarette smoke. These chemicals, on the basis of weight, account for most of the mainstream smoke. In order by prevalence, these chemicals include N2, O2, CO2, CO, nitrogen oxides, and the sulfur-containing gaseous compounds.
CO and CO2 result from the combustion of tobacco. Other than N2 and O2, CO and CO2 are the most abundant compounds in mainstream cigarette smoke, representing nearly 15 percent of the weight of the gas phase. CO2 levels (approximately 50 mg generated per cigarette) are more abundant than are CO levels (approximately 20 mg), as determined by the FTC machine-smoking method.
Nitrogen oxide gases are formed by the combustion of nitrogen-containing amino acids and proteins in the tobacco leaf (Hoffmann and Hoffmann 1997). Mainstream cigarette smoke contains mostly NO with traces of nitrogen dioxide (NO2) and nitrous oxide. The formation of nitrogen oxides is amplified by combustion with nitrate salts, and the amount formed is directly related to the nitrate concentration of the tobacco leaf (MacKown et al. 1999). The mainstream cigarette smoke contains approximately 500 μg of NO generated per cigarette. Although fresh smoke contains little NO2, the aging of the smoke converts the reactive NO to NO2, which has an estimated half-life of 10 minutes (Borland et al. 1985; Rickert et al. 1987). These gases react with water and other components in cigarette smoke to form nitrate particles and acidic constituents.
Sulfur-containing gases result from the combustion of sulfur-containing amino acids and proteins (Horton and Guerin 1974). In mainstream cigarette smoke, H2S is the most abundant of these gases (approximately 85 μg generated per cigarette), and both sulfur dioxide and carbon disulfide are present in smaller quantities (approximately 2 μg).
In addition to the volatile gases, mainstream cigarette smoke contains a wide range of volatile organic compounds (VOCs) (Counts et al. 2005; Polzin et al. 2007). The formation of these VOCs results from the incomplete combustion of tobacco during and between puffs. The generation of VOCs, as well as the previously mentioned volatile gases, is directly related to the tar delivery of the cigarette, as evidenced by machine smoking under the FTC regimen (Hoffmann and Hoffmann 1997; Polzin et al. 2007). Therefore, factors altering the yield of tar (e.g., tobacco blend, cigarette filter, filter ventilation, paper porosity, and tobacco weight) directly affect the yield of VOCs. Under certain machine-smoking conditions, the use of charcoal filters (Williamson et al. 1965; Counts et al. 2005; Laugesen and Fowles 2006; Polzin et al. 2008), variations in the temperature in the burning zone, and the presence or absence of O2 can substantially alter the levels of VOCs generated in cigarette smoke (Torikai et al. 2004). The VOCs in mainstream cigarette smoke, as a result of their high biologic activity and levels, are among the most hazardous chemicals in cigarette smoke (Fowles and Dybing 2003; IARC 2004). In developed countries, the combined exposure of smokers to mainstream cigarette smoke and nonsmokers to secondhand smoke constitutes a significant portion of the population's total exposure to certain VOCs. For example, more than one-half of the U.S. population's exposure to benzene is from cigarette smoking (U.S. Department of Health and Human Services [USDHHS] 2002). The roughly 500 VOCs in the gas phase of mainstream cigarette smoke can be subclassified by structure. Among the most significant classes are the aromatic hydrocarbons, carbonyls, aliphatic hydrocarbons, and nitriles. Although other classes of volatile compounds (e.g., acids and bases) are present, these four classes of VOCs have been the most widely studied, because of their biologic activity and overall higher levels.
Aromatics are a class of compounds defined by their structural similarity to benzene. These compounds result from incomplete combustion of the organic matter of the cigarette, most notably sugars and cellulose (Chortyk and Schlotzhauer 1973). The most abundant aromatic compounds in mainstream smoke generated from full-flavored cigarettes with use of the FTC/ISO smoking regimen are toluene (approximately 5 to 80 μg generated per cigarette), benzene (approximately 4 to 60 μg), total xylenes (approximately 2 to 20 μg), styrene (approximately 0.5 to 10 μg), and ethylbenzene (approximately 1 to 8 μg) (Counts et al. 2005; Polzin et al. 2007).
Carbonyl compounds include the ketones and aldehydes. These compounds are studied because of their reactivity and levels, which approach 1 mg generated per cigarette. The most prevalent aldehydes in mainstream smoke from cigarettes, generated using the ISO regimen, are acetaldehyde (approximately 30 to 650 μg generated per cigarette), acrolein (approximately 2.5 to 60 μg), and formaldehyde (approximately 2 to 50 μg) (Counts et al. 2005). The most prevalent ketones in mainstream cigarette smoke, generated by using the FTC/ISO smoking regimen, are acetone (approximately 50 to 550 μg generated per cigarette) and 2-butanone (approximately 10 to 130 μg) (Counts et al. 2005; Polzin et al. 2007). Spincer and Chard (1971) identified formaldehyde in both the particulate and gas phases of tobacco smoke and found that much of the formaldehyde was associated with total particulate matter (TPM). These investigators determined that formaldehyde delivery was higher in smoke from bright tobacco than in that from burley tobacco.
On the basis of total mass, hydrocarbons represent the largest VOC class in mainstream cigarette smoke (Hoffmann and Hoffmann 1997). Both saturated hydrocarbons and olefins result from the incomplete combustion of cigarette tobacco. The most abundant hydrocarbons in cigarette smoke are methane, ethane, and propane, which represent nearly 1 percent of the total cigarette effluent. Unsaturated hydrocarbons are also present in significant quantities in mainstream cigarette smoke, as evidenced by using the ISO regimen, but the olefins isoprene (approximately 70 to 480 μg generated per cigarette) and 1,3-butadiene (approximately 6.5 to 55 μg) are the most abundant unsaturated hydrocarbons (Counts et al. 2005).
The volatile nitriles, which include compounds such as HCN, acetonitrile, and acrylonitrile, are important because of their toxic effects. The most abundant nitriles in mainstream smoke generated from cigarettes by using the ISO regimen are HCN (approximately 3 to 200 μg generated per cigarette), acetonitrile (approximately 100 μg), and acrylonitrile (approximately 1 to 12 μg) (Counts et al. 2005).
In summary, cigarette smoke is composed primarily of gaseous and volatile compounds. Thus, levels of these compounds are critical in determining the overall toxicity of tobacco smoke. Differences in the design of the cigarette can have a substantial effect on the levels determined in smoke, which makes the reproducibility of results challenging, but provides knowledge of possible mechanisms to reduce the exposure of smokers.
Heavy Metals
Metals and metalloids are among the many substances contained in tobacco smoke; they are often loosely called "heavy metals" without regard to whether they are light- or heavy-mass metals or metalloids. Their chemical properties span a wide range. These substances are found as pure metals or as metals naturally associated or chemically bound to other elements that can significantly alter the chemical properties of the metals.
Although metals can be deposited on tobacco leaves from particles in the air and some fungicides and pesticides containing toxic metals have been sprayed on tobacco leaves or soils in the past (Frank et al. 1977), most of the metals present in plants are absorbed from the soil (Schwartz and Hecking 1991; Cheng 2003; Xiao et al. 2004a,b). Soils, therefore, including any amendments to the soil, such as sludge, fertilizers, or irrigation with polluted water have been the predominant source of metals found in tobacco grown in various geographic areas (Bache et al. 1985; Mulchi et al. 1987, 1991, 1992; Adamu et al. 1989; Bell et al. 1992; Rickert and Kaiserman 1994; Stephens et al. 2005). Cadmium and lead content in tobacco and smoke have been correlated with the content in the soil in which the tobacco was grown, after adjustment for the amendments to the soil (Bache et al. 1985; Adamu et al. 1989; Mulchi et al. 1991, 1992; Bell et al. 1992; Rickert and Kaiserman 1994; Stephens et al. 2005). In addition, Rickert and Kaiserman (1994) showed that heavy metals in the air can be important. For example, significant changes in the lead concentrations in the air between 1974 and 1988 accounted for most of the changes in lead levels in tobacco during that period. Researchers have associated the mercury content in tobacco with environmental factors and soil in geographic areas where the tobacco was grown (Rickert and Kaiserman 1994). Mulchi and colleagues (1992) have also suggested that consideration of soil pH is important to understanding the relationship between metals in the soil and metals in the tobacco leaf. Because of differences in the soil, air, and metal uptake by the tobacco plant, the metal content of tobaccos varies widely.
Most metals and metalloids are not volatile at room temperature. Pure metallic mercury is volatile, but only a few forms are volatile at temperatures lower than 100ºC. The temperature of tobacco that burns at the tip of a cigarette may reach 900ºC (Baker 1981). A burning cigarette tip is hot enough to volatilize many metals into the gas phase, but by the time the smoke is inhaled or rises in a plume from the cigarette as secondhand smoke, most of the metals have condensed and moved into the particulate portion of the smoke aerosol (Baker 1981; Chang et al. 2003).
The range of levels of toxic metals found in tobacco smoke reflects differences in cigarette manufacturing processes, ventilation, additives, concentrations in the tobacco, and the efficiency with which the metal transfers from the leaf to the smoke. The transfer rate of metals from tobacco into smoke also depends on the properties of the metal (Krivan et al. 1994). Because tobacco plants easily absorb and accumulate cadmium from the soil, cadmium is found at relatively high concentrations in tobacco leaves. This accumulation, along with the high percentage of transfer from the leaves into the smoke (Schneider and Krivan 1993), yields high cadmium levels in tobacco smoke (Chiba and Masironi 1992). Kalcher and colleagues (1993) developed a model for the behavior of metals in mainstream smoke and found that most of the cadmium in tobacco smoke is in the particulate phase, whereas lead is equally partitioned between the particulate and gas phases. Cadmium levels have been reported to range from 10 to 250 ng generated per cigarette in the particulate phase (Allen and Vickroy 1976; Bache et al. 1985; Nitsch et al. 1991; Schneider and Krivan 1993; Krivan et al. 1994; Rhoades and White 1997; Csalári and Szántai 2002; Torrence et al. 2002) to a lower level of 1 to 31 ng in the gas phase (Nitsch et al. 1991). More recent studies of cadmium levels in particulate matter in smoke from commercial cigarettes smoked under FTC/ISO conditions reported a range of 1.6 to 101.0 ng generated per cigarette (Counts et al. 2005; Pappas et al. 2006). Not surprisingly, Counts et al. (2005) also showed that levels of cadmium in smoke generated using more intense smoking regimens such as MDPH (12.7 to 178.3 ng generated per cigarette) and CAN (43.5 to 197.1 ng generated per cigarette) were higher than when using FTC/ISO. This increase was also seen with other metals tested. These studies also demonstrated that changes in cigarette design, such as introducing filter ventilation, reduces the delivery of metals under FTC/ISO smoking conditions. In counterfeit cigarettes, levels of cadmium in particulate matter from mainstream smoke can be significantly higher, ranging from 40 to 300 ng generated per cigarette, under FTC smoking conditions (Pappas et al. 2007).
Lead also transfers well from tobacco to smoke (Schneider and Krivan 1993); measurements range from 18 to 83 ng generated per cigarette in the particulate phase (Allen and Vickroy 1976; Nitsch et al. 1991; Schneider and Krivan 1993; Krivan et al. 1994; Csalári and Szántai 2002; Torrence et al. 2002; Baker et al. 2004) and from 6 to 149 ng in the gas phase (Nitsch et al. 1991). More recent studies of lead levels in particulate matter in smoke from commercial cigarettes smoked under FTC/ISO conditions reported a range of 4 to 39 ng generated per cigarette (Counts et al. 2005; Pappas et al. 2006). Studies of cigarettes in the United Kingdom have documented concentrations of heavy metals in a number of counterfeit cigarette brands that were higher than those in domestic products (Stephens et al. 2005). These metals included arsenic, cadmium, and lead. In counterfeit cigarettes, levels of lead in mainstream cigarette smoke can be significantly higher, ranging up to 330 ng generated per cigarette, under FTC smoking conditions (Pappas et al. 2007). Studies have also found similar levels of nickel in both phases: particulate levels range from 1.1 to 78.5 ng generated per cigarette (Bache et al. 1985; Nitsch et al. 1991; Schneider and Krivan 1993; Torjussen et al. 2003), and gas phase levels range from 3 to 57 ng (Nitsch et al. 1991).
Tobacco smoke also contains lower levels of other metals. The range of levels found in the particulate phase includes cobalt, 0.012 to 48.0 ng generated per cigarette; arsenic, 1.5 to 21.0 ng; chromium, 1.1 to 1.7 ng; antimony, 0.10 to 0.13 ng; thallium, 0.6 to 2.4 ng; and mercury, 0.46 to 6.5 ng (Allen and Vickroy 1976; Suzuki et al. 1976; Nitsch et al. 1991; Schneider and Krivan 1993; Krivan et al. 1994; Rhoades and White 1997; Milnerowicz et al. 2000; Shaikh et al. 2002; Torrence et al. 2002; Baker et al. 2004; Pappas et al. 2006). Gas phase levels depend on the volatility of the metals or metal complexes. Cobalt levels range from less than 1 to 10 ng generated per cigarette, and mercury levels range from 5.0 to 7.4 ng generated per cigarette (Nitsch et al. 1991; Chang et al. 2002). In a limited analysis, Chang and colleagues (2003) found arsenic and antimony in the gas phase but did not provide quantitative results.
Studies have identified radioactive elements in tobacco and tobacco smoke. Lead 210, a product of radioactive decay of radon, was found in tobacco (Peres and Hiromoto 2002) and is transported at low levels in tobacco smoke (Skwarzec et al. 2001). Most of the lead in tobacco smoke is the nonradioactive isotopes. Polonium, an element found only in radioactive forms, is also a product of radioactive decay of radon. Some researchers have found polonium 210 in tobacco (Skwarzec et al. 2001; Peres and Hiromoto 2002; Khater 2004), and others estimated transfer of 11 to 30 percent of the amount in tobacco to tobacco smoke (Ferri and Baratta 1966). The presence of a filter and the type of filter used can alter the amount of polonium transferred into mainstream smoke; some filters remove 33 to 50 percent of the polonium from the smoke (Ferri and Baratta 1966).
In summary, the levels of metals in tobacco smoke are primarily a function of their content in the soil in which the tobacco is grown, added substances such as fertilizer, and the design of the cigarette. Study findings indicate that (1) growing conditions for tobacco contribute to the levels of metals in cigarettes manufactured worldwide and (2) some counterfeit cigarettes have higher levels of metals than do domestic commercial cigarettes. This evidence has proved that tobacco-growing conditions can alter the concentrations of metals in cigarette tobacco and therefore the levels in the smoke.
Aromatic Amines
Aromatic amines and their derivatives are used in the preparation of dyes, pharmaceuticals, pesticides, and plastics (Brougham et al. 1986; Bryant et al. 1994; Centers for Disease Control and Prevention [CDC] 1994) and in the rubber industry as antioxidants and accelerators (Parmeggiani 1983). Because of their widespread use, aromatic amines are prevalent and may be found as contaminants in some color additives, paints, food colors, and leather and textile dyes and in the fumes from heating oils and fuels. Studies that measured aromatic amines in the ambient environment detected their presence and determined concentrations in air, water, and soil (Birner and Neumann 1988; Del Santo et al. 1991; Ward et al. 1991; Skipper et al. 1994; Sabbioni and Beyerbach 1995). Aromatic amines consist of at least one hydrocarbon ring and one amine-substituted ring, but these agents have diverse chemical structures. Chemically, aromatic amines act as bases and most exist as solids at room temperature.
Some scientists have suggested that aromatic amines are present in unburned tobacco (Schmeltz and Hoffmann 1977) and are also formed as combustion products in the particulate phase of tobacco smoke (Patrianakos and Hoffmann 1979). Investigators determined levels of aromatic amines in both mainstream and sidestream smoke (Hoffmann et al. 1969; Patrianakos and Hoffmann 1979; Grimmer et al. 1987; Luceri et al. 1993; Stabbert et al. 2003a). The identified compounds include aniline; 1-, 2-, 3-, 4-toluidine; 2-, 3-, 4-ethylaniline; 2,3-, 2,4-, 2,5-, 2,6-dimethylaniline; 1-, 2-naphthylamine; 2-, 3-, 4-aminobiphenyl; and 2-methyl-1-naphthylamine. The most commonly studied compounds from this class are shown in Figure 3.6. Stabbert and colleagues (2003a) found that aromatic amines reside primarily in the particulate phase of smoke, except for the more volatile amines such as o-toluidine; only 3 percent of o-toluidine was found in the gas phase. Studies have reported that sidestream smoke contains substantially higher levels of aromatic amines than does mainstream smoke, but these levels depend on the parameters for puffing the cigarette (Patrianakos and Hoffmann 1979; Grimmer et al. 1987; Luceri et al. 1993). For mainstream smoke, the levels of aromatic amines were reported to be 200 to 1,330 ng generated per cigarette (Luceri et al. 1993; Stabbert et al. 2003a), but studies have reported much higher levels in sidestream smoke (Luceri et al. 1993). More recently, one study reported the following levels of aromatic amines in mainstream cigarette smoke (Counts et al. 2005). Using the ISO regimen, these investigators determined that levels were 3 to 27 ng generated per cigarette for 1-aminonaphthalene; 2 to 17 ng for 2-aminonaphthalene; 0.6 to 4.2 ng for 3-aminobiphenyl; and 0.5 to 3.3 ng for 4-aminobiphenyl. These levels increased on average by approximately 115 percent when the MDPH smoking regimen was used and by approximately 130 percent under the CAN smoking regimen.
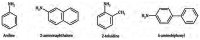
Figure 3.6
Commonly studied aromatic amines in tobacco smoke.
Levels of aromatic amines in tobacco smoke are influenced by both the chemical constituents in the tobacco and the chemical and physical processes of the burning cigarette. Levels of aromatic amines in smoke from cigarettes made with dark tobacco are higher than those in cigarettes made from light tobacco (Luceri et al. 1993). For typical U.S.-blended cigarettes, there is a linear correlation between levels of aromatic amines and tar in the smoke (Stabbert et al. 2003a).
Sources of nitrogen in the tobacco also significantly influence levels of aromatic amines in tobacco smoke. Nitrate is a primary factor in altering the level of aromatic amines in tobacco smoke, and its presence is influenced by the use of nitrogen fertilizers (Patrianakos and Hoffmann 1979; Stabbert et al. 2003a). Protein in tobacco is known to be a good source of biologic nitrogen, and studies have reported that higher nitrogen content from elevated protein in tobacco increased the yields of 2-naphthylamine and 4-aminobiphenyl (Patrianakos and Hoffmann 1979; Torikai et al. 2005). Cigarette smoke from bright tobacco had lower aromatic amine levels than expected compared with the smoke of U.S. blended cigarettes, possibly because of the lower nitrogen content in bright tobacco (Stabbert et al. 2003a). Combustion temperature is also a factor in the generation of aromatic amines in tobacco smoke, because lower temperatures yielded lower levels of aromatic amines in smoke (Stabbert et al. 2003b). Other investigators have suggested that increased cellulose levels in tobacco can decrease aromatic amines in the smoke (Torikai et al. 2005), and in another study, celluloseacetate filters removed a substantial portion of aromatic amines from mainstream smoke (Luceri et al. 1993).
In summary, it appears that the nitrogen content in tobacco, either from protein levels or use of nitrogen fertilizer, is a primary determinant of aromatic amine levels in tobacco smoke. The type of tobacco used in the cigarette filler also alters these levels in tobacco smoke.
Heterocyclic Amines
Heterocyclic amines (HCAs) are a class of chemical compounds that contain at least one cyclic ring and an amine-substituted ring. HCAs act as basic compounds because of the amine functional group. HCAs can occur in food stuff, such as grilled meats, poultry, fish, and tobacco smoke (Sugimura et al. 1977; Sugimura 1997; Skog et al. 1998; Murkovic 2004). HCAs are classified in two groups: one is produced by the pyrolysis of amino acids and proteins through radical reactions, and the other is generated by heating mixtures of creatinine, sugars, and amino acids (Sugimura 1997; Murkovic 2004). The first group dominates when the pyrolysis temperature is high, whereas the second group is predominant at low temperatures commonly used to cook meat (Sugimura 1997). In tobacco smoke, the primary HCAs are 2-amino- 9H-pyrido[2,3-b]indole; 2-amino-3-methyl-9H-pyrido[2, 3-b]indole; 3-amino-1,4-dimethyl-5H-pyrido[4,3-b]indole (Trp-P-1); 3-amino-1-methyl-5H-pyrido[4,3-b]indole (Trp-P-2); 2-amino-3-methylimidazo[4,5-f]quinoline; 2-amino- 6-methyldipyrido[1,2-a:3′,2′-d]imidazole (Glu-P-1); 2-aminodipyrido[1,2-a:3′,2′-d]imidazole; and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) (Figure 3.7) (Kataoka et al. 1998).

Figure 3.7
Primary heterocyclic amines in tobacco smoke.
HCAs are not found in unburned tobacco; they are present in tobacco smoke as a result of pyrolysis and are found in the particulate phase (Manabe and Wada 1990). The chemical composition of amino acids, protein, sugars, and creatine/creatinine in the tobacco filler influences the final HCA levels in the smoke. Other components that may alter the pyrolysis of amino acids can also change HCA levels in smoke. The usual levels of HCAs in tobacco smoke were reported to be 0.3 to 260.0 ng generated per cigarette (Hoffmann et al. 2001). Manabe and Wada (1990) reported levels of 0.29 to 0.31 ng of Trp-P-1 generated per cigarette and 0.51 to 0.66 ng for Trp-P-2 in smoke condensate from five types of cigarettes. Manabe and colleagues (1991) determined an average level of 16.4 ng generated per cigarette for PhIP in tobacco smoke condensate from cigarettes purchased in Japan, the United Kingdom, and the United States.
In summary, although HCAs are not specific to tobacco products, they are found at levels in tobacco smoke particulate that must be considered when assessing the harm from the use of burned tobacco. The concentration of nitrogen-containing compounds in tobacco influences the levels of HCAs that are found in the smoke, and reducing the nitrogen content may be a means of reducing HCAs.
Effect of Additives on Tobacco Smoke
Chemical additives are introduced into cigarette tobacco for a variety of specific purposes, including pH adjustment, maintenance of moisture (humectants), amelioration of the harshness of smoke, control of the burn rate, and impartation of desirable flavor to the smoke (Penn 1997). The taste and flavor of cigarette smoke is affected primarily by the tobacco blend and is further modified with additives. Specific additives are applied to mask the harshness of lower-quality tobacco (World Tobacco 2000). Early in the processing of burley and flue-cured tobaccos, a solution called "casing" is added to the shreds of tobacco lamina. The casing is a slurry containing humectants (e.g., glycerol and propylene glycol) and flavor ingredients with low volatility (e.g., cocoa, honey, licorice, and fruit extracts) that lend a pleasant aroma. After the tobacco is aged, a top-flavoring solution is added to the finished cigarette blend. Top flavoring is generally an alcohol- or rum-based mixture containing volatile compounds (e.g., menthol) and other ingredients (e.g., aromatic compounds, essential oils, and extracts) that are added immediately before packaging (Penn 1997; Fisher 1999).
Even though the specific ingredients added to individual cigarette brands are proprietary, a collective list of 599 additives used in U.S. cigarettes has been published on the World Wide Web (Indiana Prevention Resource Center 2005). The "599 list" contains individual chemical compounds and complex additives, such as essential oils, juices, powders, oleoresins, and extracts. Included in the list are complex natural extracts and essential oils, such as anise, cassia, cedarwood, chocolate, cinnamon, ginger, lavender, licorice, nutmeg, peppermint, valerian, and vanilla. The list also includes individual organic chemical compounds, such as 1-menthol, 3-methyl pentanoic acid, anethole, β-caryophyllene, caffeine, ethyl acetate, γ-decalactone, isoamyl acetate, methyl cinnamate, sucrose, and vanillin. The compounds in the 599 list have been approved by the U.S. Food and Drug Administration as generally recognized as safe for use in foods (Hoffmann and Hoffmann 1997). Virtually any material with this approval as a food additive is used in cigarette manufacturing (World Tobacco 2000). However, this use is based on the broad assumption that additives designated as safe for ingestion are safe to burn and inhale in cigarette smoke. Because of the detoxifying action of the liver on blood coming directly from the digestive tract and the movement of blood from the lungs into the general circulation without first passing through the liver, the toxic effects associated with ingesting a compound can differ from the toxic effects of breathing it. Studies indicated that eugenol, a compound found in many natural extracts and used as an additive in clove cigarettes, had an LD50 200 times lower in Fischer rats when administered intratracheally compared with gavage (LaVoie et al. 1986). Although this did not simulate inhalation, it did raise concern about increased toxicity of this compound to the lung.
Cigarette tobacco is a complex physicochemical mixture containing several types of tobacco and numerous additives (Hoffmann and Hoffmann 1997). The flavor compounds in tobacco can be transferred into the smoke by distillation, combustion, or pyrolysis (Green et al. 1989). Newly emerging flavored "dessert" cigarettes marketed under names such as Midnight Berry, Mandarin Mint, and Mocha Taboo (Carpenter et al. 2005) may represent new sources of exposure to harmful substances, but the qualitative and quantitative differences in smoke from these cigarettes have not been described.
One of the most common tobacco additives is menthol, a monoterpene alcohol (Burdock 1995) first used in cigarettes in the mid-1920s (Reynolds 1981) and subsequently added to most cigarettes (Eccles 1994). Natural sources of menthol include plants in the mint family, namely, peppermint (Mentha piperita) and corn mint (Mentha arvensis) (Burdock 1995). Flavorants derived from natural sources generally contain a mix of compounds, in contrast to flavoring compounds that are chemically synthesized. If menthol added to the tobacco is derived from natural sources, such as peppermint, constituents such as pulegone may also be present at low concentrations. Submicrogram concentrations of pulegone (0.024 to 0.29 μg/g) were measured in 12 mentholated brands but were not detected in nonmentholated brands (Stanfill and Ashley 1999). Menthol can be added on the tobacco, the filter, or the foil pack (Wayne and Connolly 2004). Menthol levels in smoke have ranged between 0.15 and 0.58 mg generated per cigarette for several brands (Cantrell 1990). Unlike most nonmentholated cigarettes, menthol cigarettes usually contain more flue-cured and less burley tobacco, along with reconstituted tobacco made without added ammonia.
Although they generally are regarded as safe for use in foods, certain flavor-related chemicals added to cigarettes and found in cigarette smoke (Stanfill and Ashley 1999) have known toxic properties. In an analysis of 12 flavor compounds in tobacco fillers from 68 U.S. cigarette brands, concentrations of compounds were 0.0018 to 43.0 μg/g (Stanfill and Ashley 1999). Also, 62 percent of the 68 brands contained detectable levels of 1 or more of the 12 flavor compounds. Piperonal and myristicin were present at the highest concentrations. Anethole, myristicin, and safrole were found in 20 percent or more of the brands; pulegone, piperonal, and methyleugenol were each present in at least 10 percent of the brands. In four brands, safrole, myristicin, and elemicin were found together, which strongly suggests the presence of flavorings such as nutmeg or mace (Myristica fragrans) in the tobacco. Coumarin is a benzopyrone compound found in the tobacco of one menthol brand at a concentration of 0.39 μg/g. Pulegone, a monoterpene ketone found in peppermint, was present only in mentholated brands. Tentative identification of other compounds suggested the use of flavor agents such as cinnamon and ginger (Stanfill and Ashley 1999). In addition to tobacco analysis, mainstream smoke particulates from several brands were also analyzed for six flavor compounds: eugenol, isoeugenol, methyleugenol, myristicin, elemicin, and piperonal (Stanfill and Ashley 2000). Levels of these compounds in the smoke from eight U.S. cigarette brands were 0.0066 to 4.21 μg generated per cigarette. The measurements suggested that a portion of eugenol and isoeugenol in smoke from some cigarettes could be a by-product of the burning tobacco. Also, when filter ventilation holes in the cigarette were partially or fully blocked, the transfer of these compounds from tobacco filler to mainstream smoke particulates increased twofold to sevenfold.
In summary, the impact of flavor-related additives on the toxicity, carcinogenicity, and addictive properties of tobacco products has not been thoroughly studied. In addition to the known harmful properties of these compounds, they may potentiate the effects of other known smoke constituents or alter the way people smoke cigarettes. These additives may also increase the initiation and continuation of smoking in the population.
Delivery of Chemical Constituents into Tobacco Smoke
Various tobacco types are used in the manufacture of cigarettes and other tobacco products. Lamina from bright, burley, and oriental tobacco varieties, along with reconstituted tobacco sheet, is the main filler component used in cigarettes (Hoffmann and Hoffmann 1997). In addition to lamina, cigarette filler often contains puffed or expanded tobacco, tobacco stems, humectants, and various flavor additives (Hoffmann and Hoffmann 1997; Abdallah 2003a). One tobacco variety such as bright can be used, or several varieties can be mixed together in products with specific tobacco blends. Most commercial cigarettes are constructed primarily from bright tobacco or from a blend of mainly bright, burley, and oriental tobaccos, usually referred to as an American blend (Browne 1990). However, a few small geographic areas outside the United States (e.g., France) have regional preferences for cigarettes made exclusively from dark, air-cured tobacco (Akehurst 1981; Tso et al. 1982). Each type of tobacco has unique properties that influence packing density (Artho et al. 1963), burn rate (Muramatsu 1981), tar and nicotine delivery (Griest and Guerin 1977), and flavor and aroma (Davis 1976; Enzell 1976; Leffingwell 1976). Bright tobacco, also known as flue-cured or Virginia tobacco, has lower nitrogen content (i.e., less protein) and higher sugar content than do the other varieties. Burley and Maryland tobaccos are air cured and typically have higher nicotine content but reduced sugar content.
Sakuma and colleagues (1984) measured the smoke components in mainstream and sidestream smoke and found that nitrogen-containing compounds were abundant in smoke from burley tobacco, whereas the non-nitrogen-containing compounds were more abundant in smoke from bright and oriental tobaccos. Oriental tobacco is often included in blended varieties because of its unique aromatic properties (Browne 1990). Cigarettes such as light or ultralight varieties that deliver low yields of tar and nicotine by FTC/ISO machine measurement often contain puffed or expanded tobacco lamina with higher "filling power" (Kertsis and Sun 1984; Lewis 1990; Kramer 1991), which lowers the density of the tobacco rod, thus lowering the amount of tobacco in each cigarette. Several types of reconstituted tobacco sheet are also used to manufacture cigarettes (Abdallah 2003b).
Development of reconstituted tobacco was an attempt at 100-percent utilization of tobacco (Abdallah 2003b). Stems, ribs, and scrap lamina are combined with various binders and other additives to form a "reconstituted" sheet approximating the physical and chemical characteristics of a tobacco leaf (Browne 1990; Blackard 1997; Abdallah 2003b). A common additive in reconstituted tobacco is diammonium hydrogen phosphate, which is used as a pectin release agent that facilitates cross-linkage to form stable sheet material (Hind and Seligman 1967, 1969; Hind 1968). Reconstituted tobacco sheet containing this additive selectively adsorbs nicotine from surrounding lamina and enriches it in an environment abundant in ammonia precursors (Larson at al. 1980).
The stages of manufacturing a cigarette include processing the tobacco lamina and reconstituted tobacco materials and slicing them into shreds of a specific cut width. Tobacco cut widths vary from approximately 1.5 mm for a coarse cut to 0.4 mm for a fine cut (Hoffmann and Hoffmann 1997). Alternatively, the cut width may be expressed in units of cuts per inch, which range from approximately 14 to 48. Cigarettes made from fine-cut tobacco have faster static burn rates resulting in fewer puffs (Resnik et al. 1977). A consequence of using tobacco filler with a fine-cut width is that the ratio of filler surface area to void volume increases and may increase the efficiency of the tobacco column to filter large aerosol particles (Keith and Derrick 1960).
The papers used in cigarettes are generally flax or linen fiber and may contain additives (Browne 1990). Salts often are added to the cigarette paper as optical whiteners to achieve a target static burn rate and to mask the appearance of sidestream smoke (Schur and Rickards 1960; Owens 1978; Durocher 1984). A key physical property of the paper wrapper is its porosity. Papers with high porosity facilitate diffusion of gases in and out of the tobacco rod (Newsome and Keith 1965; Owen and Reynolds 1967). Volatile smoke constituents such as CO readily diffuse through a porous wrapper, so delivery to the smoker is lower than that with less volatile constituents. High-porosity papers also permit more O2 to diffuse inward, which increases the static burn rate and the air-flow through the tobacco column that dilutes the smoke. A faster-burning cigarette yields fewer puffs, reducing tar and nicotine delivery per cigarette (Durocher 1984). Porosity of the paper, filler cut width, filter efficiency, and tobacco density all make important contributions to reduction of pressure drop in the tobacco rod, which is a key index related to acceptance by smokers (Norman 1999). Smokers prefer a cigarette on which they do not have to draw too hard because of changes in pressure drop as a result of design. A separate but related parameter, filter pressure drop, is directly related to smoke delivery and filter efficiency (Norman 1999).
In 2006, cigarette lengths generally fell into one of four categories in the U.S. market: king-size filter cigarettes (79–88 mm; accounting for 62 percent of the market); long (94–101 mm; 34 percent of the market); ultra long (110–121 mm; 2 percent of the market); and regular, nonfilter cigarettes (68–72 mm; 1 percent of the market) (FTC 2009). The usual diameter of a conventional cigarette is 7.5 to 8.0 mm (Norman 1999), although some "slims" have diameters of 5 to 6 mm. The amount of tobacco consumed varies with the circumference of the cigarette, and in cigarettes with smaller circumference, delivery of constituents in the smoke to the smoker decreases accordingly (Ohlemiller et al. 1993). The greater surface of the wrapper in long cigarettes increases the opportunity for gaseous diffusion out of the cigarette, which can (1) reduce delivery of highly volatile constituents of mainstream smoke to the smoker, but increase delivery to the nonsmoker and (2) increase the static burn rate as more O2 diffuses inward (Moore and Bock 1968). However, long cigarettes generally facilitate delivery of higher tar and nicotine levels, because more tobacco mass is burned.
Before the 1950s, most cigarettes were about 70 mm long and unfiltered (Hoffmann and Hoffmann 1997). The addition of a filter tip to a cigarette can greatly reduce delivery of many chemical constituents of mainstream smoke as determined by the FTC/ISO machine-smoking method (Fordyce et al. 1961; Williamson et al. 1965). This reduction was attributed to filtering of the smoke particulate and reducing the amount of tobacco in each cigarette. Cost savings are also achieved because the filter material is less expensive than the tobacco (Browne 1990). Filters provide a firm mouthpiece and permit the smoker to avoid direct contact with the tobacco. Cigarettes with modern cellulose-acetate filter tips gained about 96 percent of the market share by the 1970s (Hoffmann and Hoffmann 1997). In the United States, cellulose-acetate filter tips are the most popular and can selectively remove certain constituents of the smoke, including phenols and alkylphenols (Hoffmann and Wynder 1963; Spears 1963; Baggett and Morie 1973; Morie et al. 1975). Typically, a bonding agent such as triacetin or glycerol triacetate is used to facilitate filter manufacturing (Browne 1990). The filtration efficiency is proportional to the length, diameter, size, and number of fiber strands and the packing density of the cigarette (Keith 1975, 1978; Eaker 1990). Flavoring agents or other materials can also be incorporated into the filter design.
Extensive research from the 1960s has examined the use of activated charcoal in the cigarette filter to efficiently remove volatile compounds (Newsome and Keith 1965; Williamson et al. 1965; Keith et al. 1966). The addition of activated charcoal significantly reduced levels of volatile compounds, such as formaldehyde, cyanide, and acrolein (Williamson et al. 1965; Spincer and Chard 1971). Charcoal filters reduced the delivery of H2S to mainstream smoke (Horton and Guerin 1974). Both cellulose-acetate and charcoal filters removed some of the volatile pyridines (Brunnemann et al. 1978). Coatings with metallic oxides were extremely efficient at removing acidic gases (Keith et al. 1966). Filter designs can also be tailored to selectively pass and not trap certain classes of targeted compounds. For instance, inclusion of alkaline materials in the filter inhibits filtration of gaseous nicotine (Browne 1990).
One key technology used to reduce FTC/ISO machine-measured tar and nicotine delivery is the inclusion of microscopic ventilation holes in the paper wrapper (Harris 1890) or the filter paper. These holes cause the mainstream smoke to become diluted with air (Norman 1974). Filter ventilation holes are usually located in one or more rings about 12 mm from the mouth end of the filter (Baker and Lewis 1997). The amount of filter ventilation ranges from about 10 percent in some full-flavored varieties to 80 percent in brands measured as having very low delivery by using the FTC smoking regimen (CDC 1997). Filter ventilation also contributes to control of the burn rate (Durocher 1984). The tiny perforations can be made by mechanical means, electrostatic sparking, or laser ablation. Paper permeability can also be used to increase air dilution, although as the cigarette is consumed, this effect becomes less important. Delivery of lower levels of the constituents of mainstream smoke, as measured under FTC machine-smoking conditions, occurs when smoke drawn through the cigarette rod mixes and is diluted with air drawn through filter ventilation holes. Under FTC machine-smoking conditions, filter ventilation is highly effective in reducing delivery of chemical constituents (Norman 1974). However, the fingers or lips of smokers may cover vent holes when they smoke cigarettes and reduce the amount of air available for dilution, which results in delivery that is higher than expected (Kozlowski et al. 1982, 1996).
Cigarette smoke is formed by (1) the condensation of chemicals formed by the combustion of tobacco, (2) pyrolysis and pyrosynthesis, and (3) distillation products that form an aerosol in the cooler region directly behind the burning coal (Browne 1990). During a puff, the coal temperature reaches 800°C to 900°C, and the temperature of the aerosol drops rapidly to slightly above room temperature as it travels down the tobacco rod (Touey and Mumpower 1957; Lendvay and Laszlo 1974). As the smoke cools, compounds with lower volatility condense first, and many of the very volatile gaseous constituents, such as CO, remain in the gas phase. The cooler tobacco rod acts as a filter itself, and some portions of the smoke condense (Dobrowsky 1960) as the smoke is drawn through the tobacco column during a puff.
Torikai and colleagues (2004) examined the influence of the temperature, the pyrolysis environment, and the pH of the tobacco leaf on the formation of a wide variety of constituents of tobacco smoke. Their findings showed that, in general, the yields of the chemical constituents in tobacco smoke that present health concerns increased as the temperature increased from 300°C to 1,000ºC, but some compounds (e.g., acrolein and formaldehyde) reached their maximum yield at 500ºC and the yield remained approximately the same at higher temperatures. The presence of O2 in the pyrolysis atmosphere increased the yield of acrolein and other volatile organic compounds but lowered the levels of cyanide, phenol, and 1-aminonaphthalene. The pH of the tobacco had a mixed effect on the levels of toxic chemicals in tobacco smoke. Levels of B[a]P, cyanide, quinoline, resorcinol, and acrylonitrile increased with a lower pH, and hydroquinone and 1-naphthylamine levels increased with higher pH. The effects of the pH and pyrolysis atmosphere combine to influence the radical reactions that generate many constituents in tobacco smoke.
In summary, design features of the cigarette have a major influence on the yield of the constituents in smoke. Altering the tobacco blend, filter type and length, cut width, paper porosity, ventilation, and chemical additives alters the levels of many constituents of smoke.
Delivery of Chemicals to Smokers
In addition to cigarette design, the major factors that influence the delivery of chemicals to smokers are characteristics of puffing (puff volume, duration, and frequency), cigarette length smoked, and blocking air dilution holes on the filter tips of ventilated cigarettes (e.g., with the mouth or fingers). Testing cigarettes by using smoking machines or smokers in a laboratory setting can elucidate how certain design factors and smoking characteristics can influence the chemical components in smoke. However, the results obtained in a laboratory cannot be directly applied to populations of smokers because many factors influence the way a person smokes each cigarette.
In a laboratory setting, Fischer and colleagues (1989a) investigated the influence of smoking parameters on the delivery of TSNAs in mainstream smoke for six cigarette brands. The research included filter-tipped cigarettes with very-low-to-medium ISO/FTC yields of constituents of smoke and unfiltered cigarettes with high and very high ISO smoke yields. The major finding was that the puff profile and duration had no remarkable influence on TSNA delivery, but puff volume and frequency significantly increased TSNA yields. The dependency of TSNA delivery on the volume of smoke emitted from one cigarette (puff volume × number of puffs) was almost linear up to a total volume of approximately 500 mL. TSNA yield was equivalent for the same total volume whether the total volume was from a change in puff volume or puff frequency. Thus, the total volume drawn through a cigarette was the main factor responsible for delivery of TSNAs in mainstream smoke.
In another study, average levels of tar, nicotine, and CO per liter of smoke and per cigarette were determined for 10 brands of cigarettes smoked under 27 machine-smoking conditions (Rickert et al. 1986). Yields per cigarette were highly variable across smoking conditions, because of differences in the total volume of smoke. The results of a simple linear regression analysis indicated that up to 95 percent of the variation in tar yield per cigarette could be explained by variation in the total volume of smoke produced per cigarette. Puffing behavior (topography), especially the interpuff interval and total smoke volume per cigarette, which were influenced by puff volume, number of puffs, and length of the cigarette smoked, were the primary determinants of blood levels of constituents of cigarette smoke (Bridges et al. 1990).
The influence of machine-smoking parameters on levels of chemical constituents measured in smoke is well illustrated in the work of Counts and colleagues (2005). This research was performed according to the ISO, MDPH, and CAN regimens described earlier. The study examined levels of 44 chemicals emitted in cigarette smoke. Not surprisingly, the more intense smoking regimens resulted in higher levels of constituents in cigarette smoke. However, in some cases, the emissions of the constituents did not maintain their relative levels as a result of different burning properties of the tobacco under different regimens and because of breakthrough in charcoal filters in the more intense smoking regimens. Because the intensity of smoking changes, the delivery of chemicals to the smoker varies and cannot be assessed by using a single smoking regimen.
In studies of 129 female and 128 male smokers of contemporary cigarettes, Melikian and colleagues (2007a,b) reported data on smoking topography and exposure to toxic substances in mainstream smoke of cigarettes that deliver a wide range of nicotine as reported by the FTC/ISO method. Exposure was determined by the delivered dose and urinary biomarkers. The first study focused on whether differences in gender and ethnicity affect delivered doses of select toxicants in mainstream cigarette smoke, as a result of differences in smoking behavior or type of cigarettes smoked (Melikian et al. 2007b). Smoking topography differed significantly between females and males. Compared with men, women drew more (13.5 versus 12.0; p = 0.001) but smaller puffs (37.6 versus 45.8 mL; p = 0.0001) of shorter duration (1.33 versus 1.48 seconds; p = 0.002). Women also smoked a smaller portion of the cigarettes (36.3-mm butts [40.2 percent of cigarette length] versus 34.3-mm butts [39.2 percent of cigarette length]; p = 0.01). Although smoke volume per cigarette did not differ between women and men (p = 0.06), the daily dose of smoke was significantly higher in men (9.3 versus 8.0 liters [L]; p = 0.02), because men consume a greater number of cigarettes per day.
When data were stratified by race, no difference was found in puffing characteristics between European American and African American female and male smokers, except that African American women and men smoked equal lengths of the cigarettes (34.5- versus 33.9-mm butts; p = 0.93). However, African Americans smoked fewer cigarettes, so the daily smoke volume was significantly higher among European American smokers (8.61 versus 7.45 L for women; 10.6 versus 7.8 L for men). The emissions of select toxicants per cigarette, as determined by use of machine-smoking regimens that mimicked each smoker, were consistently greater among male smokers than among the female smokers, and they correlated significantly with delivered smoke volume per cigarette. The geometric means of emissions of nicotine from cigarettes were 1.92 mg per cigarette for women versus 2.2 mg for men (p = 0.005). Cigarettes smoked by women yielded 139.5 ng of NNK per cigarette compared with 170.3 ng for men (p = 0.0007). B[a]P emissions were 18.0 ng per cigarette for women and 20.5 ng for men (p = 0.01). Differences between women and men in delivery of toxicants in cigarette smoke to the smoker were more profound in European Americans than in African Americans. On average, African American men's smoking behavior produced the highest emissions of select toxicants from cigarettes, and European American female smokers received the lowest amounts of toxicants.
The second study by Melikian and colleagues (2007a) investigated urinary concentrations of biomarkers in relation to levels of select toxicants in mainstream cigarette smoke, as determined by using machine-smoking regimens that mimicked the smoking behavior of each smoker. In this study of 257 smokers, the researchers determined levels of nicotine, NNK, and B[a]P in mainstream smoke and concentrations of the respective urinary metabolites: cotinine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), and 1-hydroxypyrene (1-HOP). The smokers were assigned to groups according to the FTC yield of toxic substances in the cigarettes they smoked: low yield (0.1 to 0.8 mg of nicotine generated per cigarette, medium yield (0.9 to 1.2 mg), and high yield (>1.3 mg). Concentrations of urinary metabolites, expressed per level of parent compound delivered decreased with increased smoke emissions. In smokers of low-, medium-, and high-yield cigarettes, as measured by FTC methods, the respective ratios of cotinine (nanograms per milligram of creatinine) to nicotine (milligrams per day) were 89.4, 77.8, and 57.1 (low versus high; p = 0.06). Ratios of NNAL (picomoles per milligram of creatinine) to NNK (nanograms per day) were 0.81, 0.55, and 0.57 (low versus high; p = 0.05). Ratios of 1-HOP (picograms per milligram of creatinine) to B[a]P (nanograms per day) were 1.55, 1.13, and 0.97 (low versus high; p = 0.008). Similarly, for smokers who consumed fewer than 20 cigarettes per day, the means of cotinine per unit of delivered nicotine were 3.5-fold higher than those for smokers of more than 20 cigarettes per day. Likewise, a negative correlation was observed between ratios of cotinine to nicotine and delivered doses of nicotine in subgroups of smokers who used the identical brand of cigarettes, namely a filter-tipped, vented Marlboro (r = -0.59), which is a popular brand among European Americans, and Newport (r = -0.37), a menthol-flavored cigarette without filter-tip vents that is preferred by African Americans. The researchers concluded that the intensity of smoking and the mouth levels of smoke constituents significantly affect the concentrations of urinary biomarkers of exposure and should be taken into account in evaluating human exposure to toxic substances in cigarette smoke.
Regarding the influence of cigarette type on urinary biomarkers of exposure to toxic substances in mainstream smoke, there is a slight difference in puff volume and puff frequency among smokers of low-FTC-yield versus medium-FTC-yield cigarettes, as measured under FTC conditions (Djordjevic et al. 2000). Smokers of low-FTC-yield brands drew somewhat larger puffs (48.6 versus 44.1 mL) and inhaled more smoke both per cigarette (615 versus 523 mL) and daily (9.5 versus 8.2 L). However, delivered doses of NNK and B[a]P were marginally higher among smokers of medium-yield cigarettes (NNK: 250.9 versus 186.5 ng per cigarette; B[a]P: 21.4 versus 17.9 ng). On the other hand, Hecht and colleagues (2005) found no differences in urinary biomarkers of exposure to NNK and B[a]P among smokers of regular, light, or ultralight cigarettes.
Researchers have also suggested that blocking ventilation holes during smoking can result in increased delivery of smoke constituents. For example, when puff and inhalation parameters were allowed to vary, participants took significantly more and larger puffs from cigarettes with unblocked ventilation than from those with completely blocked ventilation (Zacny et al. 1986; Sweeney et al. 1999). Hoffmann and colleagues (1983) found that blocking air-dilution holes in seven brands of commercial filter-tipped cigarettes increased nicotine yields by 69 percent, tar yields by 51 percent, and CO yields by 147 percent. Another study examined a cigarette brand with tar and nicotine yields of 4.0 and 0.4 mg, respectively, under various conditions of machine smoking intended to reflect the wide range of smoking behaviors (Rickert et al. 1983). The researchers studied three levels of five smoking parameters (butt length, puff duration, puff interval, puff volume, and ventilation occlusion) and the effects on the number of puffs and TPM, and they estimated gas phase, particulate phase, and total yields of HCN. The HCN and TPM yields varied significantly under different smoking conditions. Ventilation occlusion had the most pronounced effect, accounting for 34 percent of the response variation in TPM yields and 42 percent of the response variation in total HCN yields.
Comparison of normal lip contact during smoking, which partially blocked filter vents, and smoking through a cigarette holder, which avoided blocking, showed higher nicotine boosts with normal lip contact (Höfer et al. 1991). Exposure to other smoke constituents may vary with the degree of blocking. Sweeney and colleagues (1999) found that blocking the filter vents of cigarettes with ventilation levels of at least 66 percent led to significant increases in CO exposure. The same manipulation of filter vents in cigarettes with filter ventilation levels of 56 percent or lower appeared to have negligible consequences for CO exposure. In another report, CO exposure from completely blocked filter vents was twice as high as from the unblocked vents (8.96 versus 4.32 parts per million [ppm]) (Zacny et al. 1986). Blocking filter vents also resulted in higher CO exposure in a study by Höfer and associates (1991). Blocking filter ventilation holes is not the only element of smoking topography that influences filter efficiency. More rapid or intense puffing increases flow rates, which results in less effective filtration, because the smoke passes through the tobacco column and filter material more quickly with less opportunity for adsorption on the filter's fibers. This smoking behavior also reduced the time for highly volatile gaseous materials to diffuse outward through the cigarette's paper wrapper.
An "elastic" cigarette is one that shows low levels of tar and nicotine when it is tested on a smoking machine but can potentially yield higher levels of emissions to smokers (Kozlowski et al. 2001). When cigarettes are elastic, smokers can extract as much nicotine as they need by changing their pattern of puffing on the cigarette. Analysis of tobacco from commercial American blend cigarettes purchased in the United States in 1990 revealed that the nicotine content did not differ substantially among brands that delivered a wide range of FTC-measured yields (Kozlowski et al. 1998). This cigarette design allowed delivery of virtually any amount of nicotine, depending on puffing behavior. Because there are similar amounts of other constituents in tobacco (e.g., TSNAs, metals, nitrates, and nitrites), regardless of the FTC ranking of the cigarette brand, more intense smoking to obtain a desired dose of nicotine leads to higher exposure to carcinogens. Historically, smokers have refused to use brands designed to reduce delivery of nicotine. For example, one company experimented with a modified cigarette containing denicotinized tobacco and a tar yield of 9.3 mg generated per cigarette but a nicotine yield of only 0.08 mg, as determined by using the FTC regimen, but this product was not successfully marketed (Rickert 2000).
Not all of the smoke volume delivered in the puff is inhaled by the smoker. Some escapes during mouth holding before inhalation. The depth of inhalation may be important for some smoke constituents but not for others, which is not surprising because of the complexity of the physics related to particle size that is involved with smoking and respiration. Finally, even very brief breath holding at peak inspiration can theoretically contribute to increased diffusion of some smoke constituents across alveolar membranes, as the intra-alveolar pressure increases.
There are considerable individual differences in inhalation patterns. In one study, inhaled smoke volume was measured by tracing the smoke with an isotope of the inert gas krypton (Woodman et al. 1986). The percentage of inhaled smoke (total inhaled smoke volume per total puff volume) averaged between 46 and 85 percent among persons in the study. Neither the mean inhaled smoke volume per puff nor the total inhaled smoke volume per cigarette was significantly correlated with any of the indices for puffing.
Evidence on the importance of inhalation patterns to total smoke exposure is mixed (Woodman et al. 1986; Zacny et al. 1987; Zacny and Stitzer 1996). Variations in results may be related to the small number of persons tested and to the difficulties inherent in accurately capturing the relationship between puffing indices and total inhaled smoke. Methods used include pneumography using a mercury strain gauge, whole-body (head and arms out) plethysmography, impedance plethysmography, inductive plethysmography, and inert gas radiotracers. The method most commonly used in U.S. laboratories that study smoking is inductive plethysmography, in which chest and abdominal expansions are measured by bands applied around the rib cage and the abdomen. Significant practical limitations include difficulties in accurate calibration of the systems and adequate integration of chest and abdominal expansions, especially because men tend to have greater abdominal expansion than women do. Measurement artifacts created by movement during measurement are another limitation. Studies of the accuracy of the systems have shown fair results in adults (Zacny et al. 1987). Errors in volume measurements were typically approximately 100 mL over a large number of respiratory cycles. Unfortunately, the attributes of the systems have not been well studied for the puff-by-puff evaluation of human smoking behaviors. In addition, the most useful information will come from integrating puff analyses with inhalation parameters on a puff-by-puff basis to assess mouth holding and breath holding at peak inhalation. Studies such as those cited above have shown that mechanical testing regimens cannot mimic the way people smoke cigarettes. These findings suggest the importance of expressing the levels of toxic constituents as a ratio with nicotine or puff volume in the denominator (Rickert et al. 1985; Burns et al. 2008).
The size of particles containing chemical species can affect their retention in the lung. Cigarette smoke is an aerosol formed as the vapors generated in the pyrolysis zone cool and condense. Cigarette design has been shown to control particle-size distribution in an aerosol, so particles become easier or more difficult to inhale (Stöber 1982; Ingebrethsen 1986; McRae 1990; Wayne et al. 2008). Burning finer-cut tobacco creates an aerosol with smaller particles, which are easier to inhale. Thus, changing the filler cut width can change the distribution of particle size in the aerosol and the chemistry. Particle size is also altered by air dilution. Dilution reduces the aerosol concentration and, thus, the coagulation rate. The particle size of the smoke is increased by increasing the coagulation rate or by condensing the moisture produced during combustion onto the smoke particles. According to Ishizu and colleagues (1987), the timed average particle size (equivalent diameter) for major chemical components in tobacco smoke was 0.03 to 0.5 μm, and constituents with higher boiling points tended toward larger particle sizes. Very small particles are more likely to be retained in the lungs. The overall equivalent diameter of particles of crude tar in tobacco smoke was 0.21 μm. Nicotine was usually present in small particles (e.g., 0.08 μm). Particle size influences how fast chemicals are transferred to tissue. Particles larger than 0.3 μm are more likely than smaller particles to be absorbed in the mouth and throat than in the lungs (Wayne et al. 2008).
Accurate measurement of particle size distribution in cigarette smoke is important for estimating deposition in the lung (Anderson et al. 1989). Most earlier studies (1960–1982) reported a median diameter of 0.3 to 0.5 μm, including a few ultrafine particles (<0.1 μm). Using the electrical aerosol analyzer, Anderson and colleagues (1989) reported similar values for median diameter (0.36 to 0.4 μm) for the particles emitted in smoke from U.S. commercial filter-tipped cigarettes. But, there were also distinctly smaller particles, with a median diameter of 0.096 to 0.11 μm. This finding indicated the presence of many more ultrafine particles in the smoke than was previously recognized. It is notable that the low- and ultralow-yield filter-tipped cigarettes Merit and Carlton emitted smaller particles than did the full-flavored Marlboro cigarettes. Ultrafine particles are of toxicologic importance because their deposition in the respiratory tract was significantly higher than that of the 0.3- to 0.5-μm particles. Also, the relatively large surface-to-volume ratio of the ultrafine particles could facilitate adsorption and delivery of potentially toxic gases to the lung.
An alternative analysis of the impact of particle size on deposition in the lung suggested that growth in particle size may accelerate deposition in the respiratory tract (Martonen and Musante 2000). Because of their hygroscopicity, inhaled smoke particles may grow to several times their original diameter. This study suggested that mainstream cigarette smoke could behave aerodynamically as a large cloud (e.g., 20 μm in diameter) rather than as submicrometer constituent particles. The effect of cloud motion on deposition is pronounced. For example, an aerosol with a mass median aerodynamic diameter of 0.443 μm and a geometric standard deviation of 1.44 would have the following deposition fractions: lung, 0.14; tracheobronchial, 0.03; and pulmonary, 0.11. When cloud motion is simulated, the total deposition is concentrated in the tracheobronchial compartment, especially in the upper bronchi, and pulmonary deposition is negligible. Cloud motion produces a heterogeneous deposition resulting in increased exposure of underlying airway cells to toxic and carcinogenic substances. The deposition sites correlate with the incidence of cancers in vivo.
Although most of the smoke particles deposit in the periphery of the lung, the surface concentrations of deposited particles are not significantly greater in the periphery than in centrally located airways (Muller et al. 1990). Concentrations on the surface of the central airway are relatively independent of breathing patterns and airway geometry. This finding suggests that the effects of deposition of particles from cigarette smoke cannot be greatly reduced by changing the pattern of smoke inhalation. Efforts to manipulate particle size in smoke have been described in greater detail in a report by Wayne and colleagues (2008). Their study draws on internal tobacco company documents to assess industry consideration of the role of smoke particle size as a potential controlling influence over inhalation patterns and exposure of lungs to harmful substances. The researchers reported that tobacco manufacturers evaluated manipulation of particle size to control physical and sensory attributes of tobacco products and to reduce health hazards related to exposure to tobacco smoke. Examples of design features of tobacco products that relate to potential effects on generation of particle size and distribution of particles include puff flow rate, tobaccos and experimental blends, combustion, circumference, rod length, and ventilation (Wayne et al. 2008).
In summary, smoking behavior (puff volume, number of puffs per cigarette, and percentage of ventilation holes blocked) has a major impact on the levels of toxic, carcinogenic, and addictive compounds delivered to the smoker in cigarette smoke. The puffing patterns of smokers vary considerably from person to person. To completely understand the effect of specific harmful chemical constituents on smokers, further research is needed to explore how cigarette design and the chemical makeup of cigarettes influence use of the product.
Biomarkers
General Concepts
Accurate prediction of health risks from cigarette use is complicated by several factors, including the chemical complexity of cigarette smoke, significant variations among the dose-response relationships for the many diseases associated with exposure to cigarette smoke, qualitative and quantitative changes in the dose of cigarette smoke received by smokers throughout their smoking histories, and the long latencies between the initiation of exposure and the onset of some diseases, such as various cancers, caused by smoking cigarettes. Prediction is also hampered by the ever-changing number and types of tobacco products available to consumers, as well as fluctuations in the composition of the products (Stratton et al. 2001).
Before the term "biomarker" was coined, biomedical researchers used the appearance of unique markers such as carcinoembryonic antigens (Burtin et al. 1972) to diagnose and monitor cancer or panels of metabolic or physiological risk factors (e.g., serum cholesterol, maternal serum α-fetoprotein, and serum angiotensin-converting enzyme) to predict the clinical course of adverse effects on health. During the 1980s, the National Research Council (NRC) issued a series of reports that covered the conceptual basis for using biomarkers and reviewing biomarkers related to major organ systems and diseases (Committee on Biological Markers of the NRC 1987). In an early comprehensive discussion of biomarkers as risk assessment tools, Hattis (1986) described their value in characterizing dose-response relationships, estimating internal dose, extrapolating across species, and assessing interindividual variability (DeCaprio 1997). At about the same time, Prignot (1987) published a summary of existing chemical markers of tobacco exposure that could be used to assess individual exposure to tobacco and exposure to secondhand tobacco smoke as well as to validate successful smoking cessation.
In the framework for considering biomarkers proposed by the NRC Committee on Biological Markers (1987), a set of still useful definitions was offered. In brief, exposure involves contact with the agent of concern. Dose refers to the material that enters the body. Internal dose refers to the amount of material entering the body, and biologically effective dose refers to the amount of the agent that reaches the target site(s) within the body. Markers of health effects reflect preclinical changes short of those reached when clinical disease occurs. Markers of susceptibility are linked to increased risk on exposure.
The long latency of most diseases caused by cigarette use indicates the need for predictive markers of future risk that could identify those people already experiencing preclinical effects of smoking. However, the first widely accepted tobacco biomarkers were indicators of exposure rather than predictors of disease risk. Breath CO, saliva thiocyanate (Jaffe et al. 1981), serum thiocyanate (Foulds et al. 1968), and nicotine and nicotine metabolites (Watson 1977) were prominent in the early literature for assessing exposure to cigarette smoke, and they remain in use today.
In comparison with the framework and definitions used for exposure and dose generally, a somewhat distinct set of terms has been applied to exposure to cigarette smoke. The 2001 report, Clearing the Smoke, published by the Institute of Medicine defines a biomarker of exposure as a tobacco constituent or metabolite that is measured in a biologic fluid or tissue and has the potential to interact with a biologic macromolecule (Stratton et al. 2001). The definition notes that such biomarkers are also considered as measures of internal dose. A biomarker of a biologically effective dose is defined as the amount of a tobacco constituent or a metabolite that binds to or alters a macromolecule. A biomarker of a biologic event with the potential to lead to harm is defined as a measurement of an effect attributable to exposure, including early biologic effects; alterations in morphology, structure, or function; and clinical symptoms consistent with harm. In the more general formulation, such biomarkers constitute markers of health effects.
Validated biomarkers of tobacco exposure exist, and progress has been made in developing biomarkers of biologically effective dose. The biologically effective dose represents the net effect of metabolic activation and the rate of detoxification in a target biologic tissue or bodily fluid. Many tobacco-related toxicants and carcinogens are biologically inactive until transformed by metabolic enzymes such as cytochrome P-450s into reactive intermediates. Reactive metabolic intermediates bind to macromolecules such as DNA and protein and disrupt their normal function. Not all binding to, or alteration of, a macromolecule leads to an adverse health effect. Consequently, the amount of material bound to a target macromolecule provides only an estimate of the biologically effective dose (Stratton et al. 2001). Polymorphisms of the metabolic enzymes may modify the balance of activation and detoxification and thus the potency and response of a biomarker (Norppa 2003).
Biomarkers of biologic events with the potential to lead to harm reflect changes in a cell or in cellular macromolecules that result from exposure to tobacco. These biomarkers can range from isolated changes with or without effects on function to events that clearly lead to illness or are symptoms of illness (e.g., cough). Measurable biomarkers of biologic events with the potential to lead to harm are relatively nonspecific (Stratton et al. 2001).
Few specific biomarkers have been validated as predictors of disease development (Stratton et al. 2001), although some studies indicated that DNA and protein adduct levels are associated with cancer risk (Hecht 2003). The application of biomarkers in tobacco-related disease is described in detail throughout this report and discussed briefly here.
Biomarkers of Exposure
There are diverse biomarkers of exposure. The least intrusive measurements are of chemicals and metabolic products in the breath. Levels of exhaled CO, nitric oxide, 2,5-dimethylfuran, and volatile organic compounds (e.g., benzene and toluene) are higher in the breath of smokers than in the breath of nonsmokers (Gordon et al. 2002; IARC 2004). One study showed that volatile compounds such as benzene and 1,3-butadiene have a short residence time in the body and that their concentrations in breath were a function of the number of cigarettes smoked and the time between when the smoker takes a puff and when the breath sample is collected (Gordon et al. 2002). Saliva is another biologic material that is readily accessible and inexpensive to collect. Cotinine, a metabolite of nicotine (Bernert et al. 2000), and thiocyanate, a metabolite of cyanide (Prignot 1987), can be measured in saliva; levels of both metabolites can be used to distinguish between smokers and nonsmokers.
Urinary compounds are useful markers of the uptake and metabolic processing of constituents of cigarette smoke (IARC 2004). Urinary markers of exposure to cigarette smoke are nicotine and nicotine metabolites including cotinine; minor tobacco alkaloids such as anatabine and anabasine; 1-HOP; 1- and 2-naphthol; hydroxyphenanthrenes and phenanthrene dihydrodiols; aromatic amines; heterocyclic amines; N-nitrosoproline; and NNAL (Hoffmann and Brunnemann 1983; Jacob et al. 1999; Hecht 2002; Murphy et al. 2004), thiocyanate (Prignot 1987), acetonitrile (Pinggera et al. 2005), and methylhippuric acids (Buratti et al. 1999). Nicotine and its metabolites and NNAL are specific to tobacco exposure, and compounds such as thiocyanate and 1-HOP reflect environmental sources of exposure including diet (Van Rooij et al. 1994; Sithisarankul et al. 1997; Hecht et al. 2004). In one study, levels of total NNAL, total cotinine, and 1-HOP increased with the number of cigarettes smoked per day (Joseph et al. 2005). The highest rates of increase were observed at low levels of cigarette use (1 to 10 cigarettes per day), and levels in urine plateaued at 25 to 35 cigarettes per day.
Some urinary metabolites provide information on metabolic activation and detoxification, as well as the dose (Hecht 2002, 2003). Examples are trans,trans-muconic acid and S-phenylmercapturic acid (benzene metabolites), NNAL and its glucuronides (metabolites of the TSNA NNK) (Melikian et al. 1993, 1994; Hecht 2002, 2003), and 1-HOP (a pyrene metabolite) (Hecht et al. 2004). Studies reported that concentrations of urinary 1-HOP glucuronide (Sithisarankul et al. 1997) and total 1-HOP (free and conjugated) (Van Rooij et al. 1994) correlated well with the number of cigarettes smoked per day. In one study, there appeared to be no significant difference in the urinary concentration of 1-HOP glucuronide in smokers of cigarettes containing blond (flue-cured) tobacco versus smokers of black (air-cured) tobacco (Sithisarankul et al. 1997). Other studies found that in most smokers, more than 80 percent of the nicotine dose received was accounted for by urine content of nicotine, nicotine glucuronide, cotinine, cotinine glucuronide, and trans-3′-hydroxycotinine (Benowitz et al. 1994; Davis and Curvall 1999). Total cotinine (free and conjugated) is considered the most reliable urinary marker of nicotine exposure (Murphy et al. 2004).
Examination of the blood of smokers shows elevated carboxyhemoglobin, thiocyanate, cadmium, acetonitrile, 2,5-dimethylfuran, VOCs (e.g., benzene, toluene, and styrene), the presence of nicotine and its metabolite cotinine, and NNAL (Ashley et al. 1996; Houeto et al. 1997; IARC 2004). In addition, investigators found a positive correlation between carboxyhemoglobin and exhaled CO for several hours after smoking (Hopkins et al. 1984), and serum cotinine and blood cadmium levels correlated with the number of cigarettes smoked per day (Telišman et al. 1997; Caraballo et al. 1998). The correlation between acetonitrile concentrations and the number of cigarettes smoked per day was shown to be weak (Houeto et al. 1997).
Markers of tobacco smoke exposure that were measured in other biologic tissues include PAH compounds in lung tissue, B[a]P and TSNAs in cervical mucus (IARC 2004), and TSNAs in pancreatic juice (Prokopczyk et al. 2002). Also, researchers observed that pregnant smokers had higher placental levels of cadmium than did pregnant women who did not smoke (Ronco et al. 2005a,b). In another study, cadmium was detected in the seminal fluid of smokers at higher levels than in that of nonsmokers, and the levels correlated with the number of cigarettes smoked per day (Telišman et al. 1997).
Biomarkers of Biologically Effective Dose
For cancer, a common assessment of the biologically effective dose is measurement of levels of carcinogen-DNA adducts. Strong data from animal experiments and some human studies indicate relationships among the levels of constituents of tobacco smoke, formation of carcinogen-DNA adducts, and cancer risk (Stratton et al. 2001). Levels of DNA adducts potentially provide the most direct measure of tobacco-induced DNA damage, and many studies reported higher levels in the tissues of smokers than in those of nonsmokers (Hecht 2003). In one study, most cancers causally associated with tobacco smoking showed positive evidence of increased adduct levels (Phillips 2005). However, human data on adduct formation suggested that saturation may occur at high levels of exposure (i.e., >20 cigarettes per day), causing the dose-response curve to plateau and reducing the proportional relationship between exposure and adduct levels (Godschalk et al. 2003). Little is known about the temporal variability of DNA adducts within a target or surrogate tissue. One investigator reported that levels of carcinogen-DNA adducts are indicators of carcinogenic hazards but not of quantifiable risks (Phillips 2005).
Carcinogen-DNA adducts can be measured in target or surrogate tissues. For example, they were measured in human lung tissue, exfoliated bladder cells, oral mucosa, exfoliated oral cells, and cervical cells—all sites of tobacco-derived cancers—and in surrogate tissues (e.g., carcinogen–peripheral blood lymphocyte DNA adducts) (Mancini et al. 1999; Romano et al. 1999; Stratton et al. 2001). The assumption that levels of DNA adducts in a surrogate tissue or cell reflect those in a target tissue has principally been supported by studies of animals treated with single carcinogens, but results in human biomonitoring studies have been mixed (Phillips 2005).
Additional biomarkers of biologically effective dose are (1) protein adducts, in that most carcinogen metabolites that react with DNA also react with proteins, and (2) oxidized damage to DNA bases. Protein adducts present at higher levels in smokers than in nonsmokers include hemoglobin adducts of TSNAs, 3-aminobiphenyl, 4-aminobiphenyl, o-toluidine, p-toluidine, and 2,4-dimethyl-aniline, as well as adducts from ethylation or methylation of the N-terminal valine of hemoglobin (Branner et al. 1998; Thier et al. 2001; Hecht 2003). The lung tissues of smokers have higher levels of acrolein-derived DNA lesions, one of which was identified as the mutagenic guanine adduct α-hydroxy-2′-deoxyguanosine. This lesion blocks DNA replication, potentially leading to G→T and G→A base substitution mutations (Yang et al. 2002; Zhang et al. 2007; Zaliznyak et al. 2009). The repair products of oxidative DNA lesions are water soluble and are generally excreted into urine without further metabolism. Because of extensive and rapid DNA repair, urine excretion of the oxidative DNA repair lesions reflects the average rate of oxidative DNA damage in all the cells in the body (Loft and Poulsen 1998). Levels of 8-hydroxy-2′-deoxyguanosine (8-OH-dG) (Gackowski et al. 2003) and 8-nitroguanine (Hsieh et al. 2002), both shown to indicate oxidative DNA damage, were found to be higher in the DNA of leukocytes of smokers than in those of non-smokers. Tobacco smoking was consistently shown to increase the urinary excretion rate of 8-OH-dG by 30 to 50 percent, and levels in urine decreased after smoking cessation (Loft and Poulsen 1998). In addition, both healthy smokers and smokers with cancer had urine levels of 8-hydroxyguanine that were higher than those in healthy nonsmokers (Gackowski et al. 2003). The oxidatively modified DNA base, 8-hydroxyguanine, is also a marker of oxidative stress. There is no epidemiologic evidence that high levels of oxidative DNA modification in tissue or high levels of oxidatively modified nucleic acid products in urine are predictors of cancer development in humans (Poulsen 2005).
Many mutagens and carcinogens are metabolically activated in vivo to electrophilic forms capable of interaction with cellular macromolecules (van Doorn et al. 1981). One of the mechanisms used by an organism to combat electrophilic attack is conjugation of the reactive chemical moiety with reduced glutathione, a nucleophile. This reaction causes an increase in more polar thioether conjugates, which are excreted from the body in urine and bile. Urinary thioether concentrations are used as a non-specific indicator of exposure to alkylating agents. Cigarette smoking was found to cause a dose-related increase in the urinary excretion of thioethers. Chemicals present in cigarette smoke and excreted in urine as thioethers include benzene, styrene, and vinyl chloride (van Doorn et al. 1981; Goldstein and Faletto 1993; Fisher 2000b). Increased concentrations of alkyladenines and alkylgua-nines from the reaction of alkylating agents with DNA were also observed in the urine of smokers (Hecht 2002). All three types of carcinogen biomarkers (thioethers, alkyladenines, and alkylguanines) reflect chemical uptake and the balance between activation and detoxification (Hecht 2003).
Biomarkers of Biologic Events with the Potential to Lead to Harm
Stratton and colleagues (2001) have reviewed a large number of biomarkers of biologic events with the potential to lead to harm. This review and more recent publications are summarized here. On an organ or system level, signs and symptoms of potential biologic events with the potential to lead to harm include osteoporosis, cough, hyperplasia, dysplasia, abnormal serum lipid concentrations, alterations in levels of blood coagulants, periodontal disease, and abnormal results for glucose tolerance tests (Stratton et al. 2001). On a molecular level, relevant measurements in target tissues of smokers include changes in RNA or protein expression, somatic mutations or loss of heterozygosity, alterations in promoter methylation, and mitochondrial mutations. In surrogate tissues, bio-markers of biologic events with the potential to lead to harm among smokers include leukocytosis, HPRT mutations, chromosomal aberrations, and changes in circulating lymphocytes.
Studies have identified biomarkers of biologic events with the potential to lead to harm related to cigarette smoking that are addressed in this Surgeon General's report. For example, a significant association and a dose-response relationship were shown for chromosomal aberrations induced by B[a]P diol epoxide at locus 3p21.3 in peripheral blood lymphocytes and for risk of squamous cell carcinoma of the head and neck (Zhu et al. 2002). Also, study findings suggested the frequency of promoter methylation in tumor-suppressor genes (P14, P16, P53) as a biomarker for risk of non-small-cell lung cancer among current and former smokers and cervical squamous cell cancer among smokers (Jarmalaite et al. 2003; Lea et al. 2004).
Cigarette smoking is a risk factor for bladder cancer. The increased mutagenicity of smokers' urine was first shown in 1977 by testing the brand XAD/acetone-extractable organics from urine in the Salmonella (Ames test) mutagenicity assay (Yamasaki and Ames 1977). Studies using essentially the same methods confirmed this observation (DeMarini 2004). Peak mutagenic activity of the urine occurs 4 to 5 hours after the start of smoking and decreases to pre-smoking levels in approximately 12 to 18 hours (Kado et al. 1985). Findings suggested that the mutagens are absorbed rapidly (in 3 to 5 hours).
Urinary mutagenicity generally correlates with the number of cigarettes smoked, and the level of urinary mutagenicity was found to be similar regardless of the tar level of the cigarettes smoked (Tuomisto et al. 1986; Kuenemann-Migeot et al. 1996). However, the urine from smokers of black tobacco was reported to be twice as mutagenic as that from smokers of blond tobacco, which correlated with the known increased risk for bladder cancer among smokers of black versus blond tobacco (Malaveille et al. 1989). In addition, smoking-associated urinary mutagenicity correlated with external measures of exposure (e.g., daily intake of chemicals from tobacco smoke) and with internal measures of exposure (e.g., urinary 1-pyrenol) (Pavanello et al. 2002).
Aromatic amines, heterocyclic amines, and PAHs appear to be the chemicals responsible for smoking-related urinary mutagenicity, as detected in the Salmonella assay (IARC 2004). Studies showed that urinary mutagenicity correlated with the levels of a 4-aminobiphe-nyl-DNA adduct in exfoliated urothelial cells from smokers (Talaska et al. 1991). Chemical analyses of urine from smokers with exceptionally high urinary mutagenicity revealed the presence of the mutagen 2-amino-7-naphthol, a metabolite of the bladder carcinogen 2-aminonaphthalene (β-naphthylamine) (Connor et al. 1983).
Although studies have described several biomarkers for risk of cardiovascular disease, no biomarker was specific to cigarette smoking. These biomarkers include changes in blood lipid concentrations, urine thromboxane A2 metabolites, blood F2-isoprostanes, vascular cell adhesion molecule-1, reduced platelet survival, atherosclerosis or calcium formation, and possibly elevated blood pressure (Stratton et al. 2001; Cavusoglu et al. 2004; Morrow 2005).
Symptoms and signs of biologic events with the potential to lead to harm to the respiratory system include late-occurring symptoms (cough, chronic phlegm production, wheeze, and shortness of breath) and decrements in pulmonary function, such as a notable decline in forced expiratory volume in one second (Carrell 1984; Ogushi et al. 1991; Stratton et al. 2001). Other biomarkers of biologic events with the potential to lead to harm are declines in alveolar neutrophil and macrophage counts and declines in neutrophil elastase α1-antiprotease complexes.
Some of the general markers described here can be considered as biomarkers of potential reproductive or developmental effects from maternal cigarette smoking during pregnancy. Findings in one study indicated that increased levels of F2-isoprostane in cord blood may serve as a biomarker of oxidative stress (Obwegeser et al. 1999). Another study reported biomarkers in cord blood of offspring of women who smoked during pregnancy and in maternal blood (İşcan et al. 1997). The markers included reduced levels of high-density lipoprotein cholesterol (HDLc) and apolipoprotein A-I (APO A-I) and elevated ratios of total cholesterol to HDLc, low-density lipoprotein cholesterol (LDLc) to HDLc, and APO B to APO A-I. Proteomics allows study of changes to proteins following environmental exposures. A recent comparison of up- and downregulated proteins in blood cord sera from the offspring of women who smoked during pregnancy with that of offspring of nonsmokers suggests that infants exposed in utero undergo changes in protein expression similar to those of smoke-exposed adults and animal models (Colquhoun et al. 2009). Among the changes were markers of inflammation (α2-macroglobulin), altered lipid metabolism (APO A-I), and α-fetoprotein, which is associated with fetal growth retardation (Colquhoun et al. 2009). These findings indicate that serum and cord blood lipid panels may provide biomarkers of biologic events with the potential to lead to harm to fetal metabolism of lipids.
Smoking interferes with absorption of vitamins B6, B12, and C and folic acid (Cogswell et al. 2003). Study findings indicate that lower plasma concentrations of vitamins (folate and B12) and nitric oxide from maternal smoking may result in hyperhomocysteinemia in pregnant women, a known risk factor for pregnancy-induced hypertension, abruptio placentae, and intrauterine growth restriction (Obwegeser et al. 1999; Özerol et al. 2004; Steegers-Theunissen et al. 2004). Women who smoke during pregnancy have an increased risk of delivering a low-birth-weight infant (USDHHS 2004). Decreases in birth weight were dose related to the number of cigarettes smoked (Abel 1980). Researchers reported that low concentrations of maternal serum folate and vitamin B12 were associated with higher risk of preterm delivery and low birth weight, and low-birth-weight infants had significantly lower concentrations of vitamins A, B2, E, and folate (Navarro et al. 1984; Fréry et al. 1992; Scholl et al. 1996). In other studies, placental cadmium levels were strongly correlated with birth weight in newborns of mothers who smoked (Ronco et al. 2005a). Cotinine concentrations in maternal serum and urine were also useful in predicting birth weight (Stratton et al. 2001).
In summary, several biomarkers provide an accurate assessment of exposure to toxic chemicals in cigarette smoke. Still to be determined is how accurately they can characterize differences in exposure between tobacco-burning cigarettes and the variety of potentially reduced-exposure products introduced into the market during the last few years. Biomarkers of biologically effective doses for mutagenic and carcinogenic chemicals can provide an estimate of the interaction between chemicals in smoke and target biologic tissues or bodily fluids. Genetic polymorphisms of the enzymes involved in the metabolic activation of the chemicals may influence the net balance of activation and detoxification in a target biologic tissue and complicate interpretation of the dose-response relationship between exposure and binding with macromolecular targets. Despite the large number of biomarkers of biologic events with the potential to lead to harm, most are not specific to exposure to cigarette smoke and require additional testing to establish their specificity, sensitivity, and reliability when smoking behaviors or product characteristics vary. In addition, not all biomarkers of biologic events with the potential to lead to harm may be sufficient for determining population-level effects of the product.
Genotoxicity
Cigarette Smoke Condensate
Condensate from cigarette smoke is mutagenic in a variety of systems (DeMarini 1983, 2004; IARC 1986, 2004). Most studies have used condensate generated from the smoke of reference cigarettes such as those available from the University of Kentucky, Lexington, Kentucky. Researchers using the bacterial Salmonella mutagenicity assay reported that the average mutagenicity of cigarette smoke condensates prepared from the mainstream smoke from U.S. commercial cigarettes and K1R4F reference cigarettes was not significantly different among cigarettes representing more than 70 percent of the U.S. market (Steele et al. 1995). These findings suggested that such reference cigarettes are acceptable standards for comparative mutagenicity of condensates from cigarettes purchased typically in the United States. The genotoxicity of 10 cigarette smoke condensate samples from a diverse set of cigarettes (including the K2R4F reference cigarette) and produced under different smoking-machine conditions was studied in four short-term assays: the Salmonella mutagenicity assay in frameshift strains TA98 and YG1041, the micronucleus and comet assays in L5178YTk ± 7.3.2C mouse lymphoma cells, and an assay for chromosomal aberrations in CHO-K1 cells (DeMarini et al. 2008). All 10 condensate samples were mutagenic in both strains of Salmonella and induced micronuclei, and 9 samples induced DNA damage or chromosome aberrations. While their mutagenic potencies in Salmonella spanned 7-fold when expressed as revertants per gram of condensate, they spanned 158-fold when expressed as revertants per milligram of nicotine. The range of genotoxic potencies of the condensates in the other assays was similar regardless of how the data were expressed. The overall conclusion was that there was no relation among the genotoxic potencies of the cigarette smoke condensates across the assays (DeMarini et al. 2008).
Several lines of evidence indicated that the primary sources of mutagenic activity detected in the Salmonella mutagenicity assay are aromatic amines and heterocyclic amine protein pyrolysate products (IARC 1986). Most of this activity resides in the basic or base/neutral fraction of the condensates, which contains the aromatic and heterocyclic amines. At the molecular level, the mutation spectrum of cigarette smoke condensate in the Salmonella frameshift strain TA98 was identical to that of the heterocyclic amine Glu-P-1 (DeMarini et al. 1995). The finding suggested that this class of compounds is responsible for most of the frameshift mutagenic activity of cigarette smoke condensate detected in TA98. A frameshift mutation is the insertion into or deletion from DNA of a number of nucleotides that are not three or multiples of three. In contrast, most of the mutations induced by cigarette smoke condensate in the base-substitution strain TA100 were shown to be transversions of GC→TA (78 percent), which resembled most closely the mutation spectrum of B[a]P, the model PAH (DeMarini et al. 1995). The GC→TA transversions, a common class of base substitutions found in lung tumors of smokers, were also induced by cigarette smoke condensate at the HPRT locus in human B-lym-phoblastoid MCL-5 cells (Krause et al. 1999).
Study findings indicated that most of the ability of cigarette smoke condensate to induce sister chromatid exchange (SCE) in mammalian cells may reside in the neutral and acidic/neutral fractions, suggesting that this activity is attributable to PAHs and acidic compounds, such as catechol, hydroquinone, alkylphenols, and benzaldehyde (Jansson et al. 1988).
Nicotine and its metabolites were not mutagenic in Salmonella and did not induce SCEs in mammalian cells in culture, and nicotine did not produce mutagenic urine in rats (Doolittle et al. 1995). Burning tobacco produced mutagenic chemicals, and cigarette smoke condensate contained a variety of agents exhibiting a wide range of toxic effects. Varying the amounts of 300 to 400 ingredients added to typical commercially blended test cigarettes did not alter the inherent mutagenicity or cytotoxicity of the resulting condensates or the toxic effects of inhalation of the smoke of the resulting cigarettes (Carmines 2002; Baker et al. 2004). Many of the pyrolysis products from the cigarette ingredients identified as "biologically active" were volatile compounds (e.g., benzene, toluene, and styrene) (Baker et al. 2004) and would presumably reside primarily in the gas phase of the cigarette smoke rather than in the condensate used in most in vitro assays.
DNA Damage
Many studies have demonstrated that cigarette smoke and its condensate can produce DNA strand breaks in rodents, in mammalian cells in culture, or in DNA in vitro (IARC 2004). Collectively, results of these studies are consistent with the demonstrated clastogenicity (chromosome-breaking ability) of cigarette smoke and condensate and cigarette smoke in experimental systems and in humans. Several of these studies (IARC 2004) indicated that reactive oxygen or nitrogen species may be the primary cause of the breaks in DNA strands.
Cytogenetic Effects in Rodents
Exposure of rodents to cigarette smoke by inhalation has generally produced an increased frequency of SCE in the bone marrow (IARC 1986). However, such exposure produced some negative studies and one positive study of induction of chromosomal aberrations in lung cells (DeMarini 2004). Nonetheless, this exposure consistently produced micronuclei in bone marrow, peripheral blood erythrocytes, and lung cells (IARC 2004).
Transplacental Effects in Rodents
Mice born to dams exposed to cigarette smoke by inhalation during pregnancy had elevated levels of micronuclei in the liver and peripheral blood (Balansky and Blagoeva 1989), and such exposure induced SCEs in the liver of fetal mice (Karube et al. 1989). Intraperitoneal injection of pregnant Syrian golden hamsters with the tobacco carcinogen NNK also induced micronuclei in fetal liver (Alaoui-Jamali et al. 1989), and intraperitoneal injection of pregnant mice with NNK induced oxidative damage, as determined by measurement of concentrations of 8-OH-dG DNA adducts in the fetuses (Sipowicz et al. 1997).
Studies in Humans
HPRT Mutations
In general, smoking was shown to increase the frequency of HPRT mutants in peripheral blood lymphocytes by approximately 50 percent. However, the increases did not reach statistical significance in some studies, probably because of the large interindividual variability (DeMarini 2004). An increase in transversions, in particular GC→TA, was noted frequently among smokers (IARC 2004). However, some analyses found no difference in the mutation spectrum at HPRT in smokers and nonsmokers (Curry et al. 1999). GC→TA transversions are the primary class of base substitution induced by PAHs, and an excess of this class of mutation in the HPRT mutation spectrum for smokers is consistent with exposure to PAHs in cigarette smoke.
Genotoxic Effects in Reproductive Tissues and Fluids and in Children of Smokers
Lymphocytes from pregnant women who smoked either tobacco cigarettes or marijuana cigarettes had elevated frequencies of HPRT mutants, as determined by the autoradiographic HPRT assay, and analyses of cord blood indicated that lymphocytes from the newborns also had elevated frequencies of HPRT mutants (IARC 2004; DeMarini and Preston 2005). No differences in frequencies of HPRT mutants were observed in T lymphocytes from newborns of smokers compared with those from newborns of nonsmokers, as determined by the T-cell cloning assay. However, the mutation spectra for these two groups of newborns differed significantly from those for newborns of smokers who had an increase in "illegitimate" genomic deletions mediated by V(D)J recombinase. These findings suggested alteration in the HPRT mutation spectrum and possible increase in the frequency of HPRT mutant cells in newborns of mothers who smoked compared with those in newborns of mothers who did not smoke. Another study reported that in utero exposure to cigarette smoke also resulted in increases of translocation frequencies in newborns (Pluth et al. 2000). Other evidence indicated that smoking by the mother may lead to DNA strand breaks in lymphocytes of newborns (Şardaş et al. 1995). Amniocytes from mothers who smoked may show an increase in chromosomal mutations compared with those from nonsmokers (de la Chica et al. 2005); however, researchers raised concerns about this study, such as the lack of exposure assessment, the small sample size, and the fact that the chromosomal aberrations identified were of the chromatid type, which is a type that could have been formed in the petri dish during culturing and were not present in the amniotic fluid initially (DeMarini and Preston 2005).
Reviews indicated that the cervical mucus and amniotic fluid of smokers were mutagenic and that cervical epithelial cells from smokers had higher frequencies of micronuclei compared with those from nonsmokers (IARC 2004). Findings also suggested that smoking may induce chromosomal mutations and DNA damage in sperm or ova of smokers. The evidence that smoking induced oxidative damage to sperm DNA was found in elevated concentrations of 8-OH-dG in sperm DNA of smokers compared with that of nonsmokers (Shen et al. 1997). In addition, seminal fluid from infertile male smokers showed more oxidative damage than did that from infertile nonsmokers (Saleh et al. 2002). Consistent with these observations was the finding that sperm from smokers had higher concentrations of DNA strand breaks than did sperm from nonsmokers (Potts et al. 1999). Concentrations of DNA adducts in sperm, measured by the 32P-postlabeling assay were also higher among current smokers than among lifetime nonsmokers (Horak et al. 2003). Collectively, these data from studies of humans are consistent with the recent demonstration that exposure to cigarette smoke by inhalation resulted in germ-cell mutations in male mice (Yauk et al. 2007).
Cytogenetic Effects
Micronuclei. Many studies have examined the influence of smoking on the frequency of micronuclei in peripheral lymphocytes; the results were mixed (Bonassi et al. 2003). A reanalysis of pooled data from 24 databases from the Human MicroNucleus international collaborative project showed that smokers did not have an overall increase in micronuclei frequency in lymphocytes. However, a significant increase in micronucleus frequency was found in heavy smokers (i.e., those smoking 30 cigarettes or more per day) who were not exposed occupationally to genotoxic agents. Studies also found elevated micronuclei frequencies in the tracheobronchial epithelium of smokers (Lippman et al. 1990).
Sister chromatid exchange. In contrast to frequency of micronuclei, SCE frequencies in peripheral lymphocytes are generally higher among smokers than among nonsmokers. Numerous studies of SCE frequencies in peripheral lymphocytes showed that cigarette smoking induced SCEs, which can then be a confounding factor in occupational studies (IARC 2004). The findings indicated that of all the cytogenetic endpoints, SCE is the most sensitive to the effect of smoking.
Chromosomal aberrations. Studies of large populations with use of chromosome banding techniques to assess chromosomal aberrations have had mixed results. One study reported that the frequency of chromosomal aberration was not increased by smoking (Bender et al. 1988), and another reported that smoking caused a 10- to 20-percent increase in the frequency (Mutation Research 1990). Smaller studies and those using molecular cytogenetic techniques also had mixed results; in some, smoking increased the frequency of chromosomal aberrations in peripheral lymphocytes, and in others, this finding was not observed (DeMarini 2004).
Mechanistic considerations include the observation that smokers had lower concentrations of folate in red blood cells than did nonsmokers, which may play a role in the higher frequency of chromosomal aberrations detected in smokers (Chen et al. 1989). Other studies found that exposure of peripheral lymphocytes from smokers to mutagens in vitro resulted in a higher frequency of chromosomal aberrations than did similar exposure of lymphocytes from nonsmokers (IARC 2004). Collectively, findings of these studies suggested that cells of smokers, especially males, were less able to repair DNA damage and that concentrations of DNA repair enzymes, fragile sites in chromosomes, and telomeric associations could be affected by recent mutagenic exposures such as smoking (DeMarini 2004). These effects of smoking varied among individuals, and were influenced by exposures other than smoking.
A large international study showed that an elevated frequency of chromosomal aberrations in lymphocytes predicted cancer risk independently of exposure to carcinogens, including cigarette smoke (Bonassi et al. 2000). However, many studies demonstrated an association between smoking and certain genetic changes that are specific predictors of various types of tumors. For example, lymphocytes of smokers had a higher frequency of fragile sites in chromosomes and metaphases with extensive breakage, as well as overexpression of fragile sites at chromosomal breakpoints associated with cancer and oncogene sites on chromosomes (Kao-Shan et al. 1987). Smoking was associated with chromosomal instability in lymphocytes as a biomarker for predisposition to oral premalignant lesions (Wu et al. 2002). In addition, smoking was associated with mutagen sensitivity of lymphocytes as a predictor of cancer of the upper aerodigestive tract. An analysis of normal bronchial epithelium using a molecular cytogenetic technique found a significant percentage of trisomy 7 in cancer-free tobacco smokers (Lechner et al. 1997). Another study reported a significant increase in the loss of heterozygosity involving microsatellite DNA at three specific chromosomal sites containing putative tumor-suppressor genes in histologically normal bronchial epithelium from long-term smokers (Mao et al. 1997; Wistuba et al. 1997). The frequency of chromosomal aberrations was much higher in lung tumors from smokers (48 percent) than in those from nonsmokers (11 percent), suggesting that lung cancer in smokers is a result of genetic alterations distinct from those in nonsmokers (Sanchez-Cespedes et al. 2001).
Studies also associated alterations in chromosome 9 in bladder tumors with cigarette smoking, and cytogenetic changes and smoking were associated with risk for leukemia and other myelodysplastic syndromes (IARC 2004).
DNA strand breaks and oxidative damage. A review by DeMarini (2004) reported that lymphocytes, buccal cells, and urothelial cells of smokers had higher frequencies of DNA strand breaks than those in nonsmokers, as measured by the single-cell gel electrophoresis (comet) assay, which detects broken DNA that separates from whole nuclear DNA when exposed to an electric current. Oxidative damage measured by concentrations of 7-hydroxy-8-oxo-2′-deoxyguanosine (8-oxo-dG) (a marker of oxidative damage) was elevated in lymphocytes and leukocytes, urine, and lung tissue of smokers. In vitro studies, including some in human cells, also found that cigarette smoke or its components induced DNA or oxidative damage. Collectively, these studies suggested that smoking induced oxidative DNA damage.
Mutations in tumors associated with smoking. In a review of studies in 2004, IARC noted that the TP53 gene was mutated most frequently in lung tumors associated with smoking, and the details of this observation were reviewed extensively (Pfeifer et al. 2002; Pfeifer and Hainaut 2003; IARC 2004). Mutations in the TP53 gene were more common in smokers than in non-smokers, and a direct relationship existed between the frequency of TP53 mutations and the number of cigarettes smoked. TP53 mutations were found in preneoplastic lesions of the lung, indicating that they were early events linked temporally to DNA damage from smoking.
Among the mutations of the TP53 gene in lung tumors of smokers, 30 percent were GC→TA transversions, whereas only 10 percent of the TP53 mutations in lung tumors of nonsmokers or in other tumors were of this type. The sites at which these mutations occurred in the TP53 gene corresponded with the sites of DNA adducts remaining after cells were exposed to diol epoxides of PAHs and allowed to undergo a period of DNA repair (Smith et al. 2000). The mutations in the tumors were targeted at methylated CpG sites on chromosomes, and there was a bias for most of the mutated guanines of the GC→TA mutations to be on the nontranscribed DNA strand in lung tumors from smokers, which is attributable to the preferential repair of DNA adducts on the transcribed strand (Yoon et al. 2001).
Mutations in the KRAS gene (codons 12, 13, or 61) were shown to occur in approximately 30 percent of lung adenocarcinomas of smokers and are primarily GC→TA transversions, as seen in the TP53 gene (Gealy et al. 1999). As with the TP53 gene, the site at which the majority of a particular type of PAH adducts are formed in the KRAS gene (the first position of codon 12) corresponded with the position where a high frequency of GC→TA transversions occur in lung tumors associated with smoking (Tretyakova et al. 2002). Similar to TP53 mutations, KRAS mutations occurred early in carcinogenesis of the lung, and 66 percent of the mutations in the KRAS gene in smoking-associated lung tumors were GC→TA transversions (Keohavong et al. 2001).
These observations, along with substantially more data, suggest that the TP53 and KRAS mutations in lung tumors of smokers are due to the direct DNA damage resulting from the carcinogens in cigarette smoke, especially PAHs (Pfeifer and Hainaut 2003). Researchers have suggested that other factors, especially selection, may also play a role in the observed mutation spectrum in smoking-associated lung tumors (Rodin and Rodin 2005).
Cytotoxicity
Cytotoxicity refers to a specific destructive action on cells. The cytotoxicity of cigarette smoke has been shown to manifest as several pathological conditions including irritation and inflammation, cell proliferation and hyperplasia, oxidative stress and damage, and decreased organ function (Andreoli et al. 2003). Studies demonstrated the presence of cytotoxic agents in the gas and particulate phases of cigarette smoke, and HCN and acrolein were identified as specific cytotoxic agents in the gas phase (Thayer and Kensler 1964; Battista 1976a). In the particulate phase, nonvolatile and semivolatile fractions, especially semivolatile acidic and neutral fractions, were found to demonstrate cytotoxic activity (Curvall et al. 1984, 1985; Matsukura et al. 1991).
Study findings indicate that cytotoxicity may play a role in several tobacco-related chronic diseases, including emphysema, carcinogenesis, and atherosclerosis (Bombick et al. 1998; Andreoli et al. 2003). For example, injury to cells of the respiratory system by cigarette smoke is thought to be mediated by smoke-induced inflammation and damage from free radicals (Churg and Cherukupalli 1993). Thus, the usefulness of in vitro cytotoxicity tests lies in their ability to measure indicators of cellular injury that may correlate with or predict inflammation (Stratton et al. 2001).
Many early cytotoxicity studies focused on damage to ciliated organisms (paramecium), clam gill epithelium, and animal trachea (Wang 1963; Weiss and Weiss 1964; Wynder et al. 1965; Dalhamn 1970; Battista 1976a,b; Donnelly et al. 1981a,b; Curvall et al. 1984), as well as cells such as adipocytes, macrophages, and human tumor cell lines (Thayer and Kensler 1964; Thayer 1976a,b; Drath et al. 1981; Curvall et al. 1984, 1985). Ciliatoxicity assays measure the time to incapacitation of ciliated cells or the time required by ciliated respiratory cells to transport inert particles when exposed to cigarette smoke. Impaired ciliary function and mucus transport in an intact respiratory system precede metaplasia in bronchial epithelium. Assays with isolated or cultured cells typically assess inhibition of metabolic activity or cellular growth in the presence of cigarette smoke or damage to the cell membrane (Wynder and Hoffmann 1967).
Subsequent research on the cytotoxicity of cigarette smoke has frequently used the neutral red incorporation assay to evaluate smoke from different types of cigarettes or tobaccos (Bombick et al. 1997a,b, 1998; Foy et al. 2004). This assay is based on the uptake of neutral red dye into the lysosomes of viable cells. Injury to the plasma membrane or lysosomal membrane was shown to decrease uptake and retention of the dye (Babich and Borenfreund 1987). One study demonstrated that flue-cured tobacco produced smoke condensate that was significantly more cytotoxic in the neutral red incorporation assay than was condensate from burley tobacco smoke (Bombick et al. 1998). In addition, no difference was found in the cytotoxicity of smoke condensate from reference cigarettes representing commercial ultralow-tar (1R5F), low-tar (1R4F), or unfiltered full-flavored (2R1) cigarettes. In contrast, with this assay, whole mainstream smoke and the vapor phase of mainstream smoke from a 2R1 cigarette were more cytotoxic than those from a 1R4F cigarette, and those from a 1R4F cigarette were more cytotoxic than those from a 1R5F cigarette (Bombick et al. 1997a). In addition, sidestream smoke (whole smoke and vapor phase) was more cytotoxic than mainstream smoke, as determined in the neutral red incorporation assay. The same laboratory reported that neither a low-nitrogen tobacco blend with a cellulose-acetate filter (11.6 mg tar in mainstream smoke) nor a traditional U.S. tobacco blend with a charcoal filter (10.4 mg tar in mainstream smoke) reduced the cytotoxicity of the condensate of full-flavored, low-tar cigarettes in the neutral red incorporation assay (Bombick et al. 1997b).
In more recent studies, researchers reported that heating the tobacco at a low temperature instead of burning it reduced the cytotoxicity of the smoke, as determined by the neutral red incorporation assay (Tewes et al. 2003). However, the reduction was greater in the particulate phase than in the gas phase (Patskan and Reininghaus 2003). Less frequently used in vitro assays for cytotoxicity include the dye exclusion assay (Hopkin et al. 1981; Hopkin and Evans 1984); the lactate dehydrogenase release assay; the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT) uptake assay; and the kenacid blue binding assay (Putnam et al. 2002).
Smoking-machine conditions are a determinant of the cytotoxicity of cigarette smoke condensate (Foy et al. 2004; Roemer et al. 2004). Smoke condensates from U.S. commercial cigarettes ranging from very low or ultralow tar to full flavor as ranked by the FTC/ISO method, and also experimental reference cigarettes ranging from ultralow tar to low tar to full flavor, demonstrated a higher level of cytotoxicity when produced under smoking-machine conditions that generated higher smoke yields. The increase in cytotoxicity was measured in both the particulate and the gas phases expressed on a per cigarette basis. The increase in cytotoxicity measured in smoke produced under more intense smoking conditions was greatest for the particulate phase of the full-flavored commercial cigarettes and least for the ultralight varieties. This pattern was not as evident for cytotoxicity induced in the gas phase (Roemer et al. 2004).
The cytotoxicity of machine-generated mainstream smoke from the 2R1 reference cigarette to cultured mouse fibroblast L-929 cells was reduced by increasing the age of the smoke and the amounts of charcoal in an acetate filter (versus acetate alone) (Sonnenfeld et al. 1985). Investigators showed that cytotoxic effects on lung epithelial cells were attributable to oxidants and aldehydes present in the volatile phase of the smoke or formed in the cells on exposure to the smoke (Hoshino et al. 2001). In one study, selective reduction of compounds in the gas phase by an activated carbon filter decreased the cytotoxicity of the gas phase of the smoke from a commercial cigarette to lung epithelial cells (Pouli et al. 2003). (The compounds were acetaldehyde, acetone, acetonitrile, acrolein, acrylonitrile, benzene, 1,3-butadiene, 2-butanone, 2,5-dimethylfuran, ethylbenzene, furan, isobutyraldehyde, isoprene, meth-acrolein, methanol, 1,3-pentadiene, propionaldehyde, propionitrile, toluene, and m-xylene.) However, in other research, a decrease in intracellular concentrations of reduced glutathione in a human type II–like cultured lung cell line (A549) exposed to whole smoke was significantly greater than that produced by smoke filtered through a Cambridge filter pad (Ritter et al. 2004). This finding suggests that chemicals in the particulate phase of cigarette smoke produce an immediate depletion of an important cellular antioxidant. The A549 cell line has been extensively used to study human lung damage by single chemicals and complex chemical mixtures. This cell line may be more useful for studying substances that are active in their administered form, rather than for studying those that require biotransformation to reactive metabolites, because some cytochrome P-450 isoforms are not expressed in A549 cells (Castell et al. 2005).
Recent mechanistic studies identified apoptosis and necrosis as important mechanisms of cytotoxicity of cigarette smoke to cultured mammalian lung cells (Hoshino et al. 2001; Piperi et al. 2003; Pouli et al. 2003). In one study, the viability of alveolar type 2 A549 cells was reduced by smoke extract from a commercial cigarette in a time- and concentration-dependent manner, as measured by the reduction of MTT (Hoshino et al. 2001). In another study, the viability of mouse lung LA-4 cells was reduced by the gas phase of commercial cigarette smoke in a concentration-dependent manner, as measured by lactate dehydrogenase leakage and reduced metabolic activity (WST-1 assay) (Piperi et al. 2003). In both studies, apoptosis was seen at low concentrations of smoke and necrosis was seen at higher concentrations. One of the studies found that smoke extract increased intracellular oxidative activity (Hoshino et al. 2001). The other study observed a dose-dependent reduction in reduced cellular glutathione levels (Piperi et al. 2003). In addition, cells exposed to cigarette smoke showed increased protein modification (nitrotyrosine immunoreactivity) and activation of mitogen-activated protein kinase pathways. Aoshiba and colleagues (2001) reported that toxic effects on isolated alveolar macrophages from the smoke of an unfiltered commercial cigarette involved oxidative stress, an important mediator of cell death through both necrosis and apoptosis. This effect was associated with accumulation of BAX protein, mitochondrial dysfunction, and release of mitochondrial cytochrome c, but it was independent of the TP53 gene, FAS, and caspase activation. Sublethal concentrations of unfiltered extract from mainstream smoke from a commercial cigarette produced evidence of senescence in alveolar epithelial cells—A549 cells and alveolar type 2 cells isolated from normal human lungs. The senescence was characterized by dose- and time-dependent increases in β-galactosidase activity, changes in cell morphology, accumulation of lipofuscin, overexpression of the P21CIP1/WAF1/SDI1 protein, and irreversible growth arrest (Tsuji et al. 2004).
Scientists reported that the limitation of past and current in vitro tests for cytotoxicity is that the results are based on the response of single cell types or isolated tissues and do not include the influence of the whole-body system on the response (Stratton et al. 2001). However, in vitro cytotoxicity assays are useful in determining the contribution of different tobacco blends or cigarette components (e.g., the filter) to the overall cytotoxicity of the smoke and in identifying causative cytotoxic agents in smoke and mechanistic pathways. Although in vitro assays are not able to replace all conventional animal bioassays, they are increasingly seen as alternatives to animal testing of drugs and chemicals, in the European Union, the United States, and elsewhere (Höfer et al. 2004; Interagency Coordinating Committee on the Validation of Alternative Methods 2004). Many cellular pathways are activated similarly in vitro and in vivo (Devlin et al. 2005). In 2005, the Canadian government implemented a regulation requiring performance of three in vitro tests of toxicity (mutagenicity, clastogenicity, and cytotoxicity) on emissions for all cigarettes sold in Canada and that the results be reported to the Minister of Health (Canada Gazette 2005). Quantitative in vitro dose-response data could eliminate the need for use of a large number of experimental animals to achieve appropriate statistical power in an in vivo study (Parry et al. 2005).
Animal Bioassays
Researchers have tested the carcinogenic ability of tar in cigarette smoke in laboratory animals for more than nine decades and in animal inhalation studies of machine-generated cigarette smoke for more than five decades (Wynder and Hoffmann 1967). The first successful induction of cancer in a laboratory animal with a tobacco product was reported by Wynder and colleagues (1953, 1957) with the application of cigarette tar to mouse skin. They observed a clear dose-response trend between the amount of tar applied to the skin of the mice and the percentage of animals bearing skin papilloma and carcinoma in the test group. Skin-painting studies typically used condensate from cigarette smoke produced under standard FTC or ISO conditions, allowing comparisons among studies. More recent studies showed that smoking-machine conditions influence the measures of in vitro mutagenicity and cytotoxicity of smoke condensate, expressed on a per cigarette or per milligram of tar basis (Roemer et al. 2004; Rickert et al. 2007). However, skin-painting studies typically focused on product characteristics such as tobacco filler, paper, and additives rather than on smoke condensate produced under different smoking-machine conditions. One study demonstrated that tobacco blend, filter type, and flavoring materials are determinants of the composition of mainstream smoke, whereas the amount of tobacco in the cigarette, the dimensions of the cigarette, and the filter type influence smoke yield (Borgerding and Klus 2005). Future skin-painting studies will likely use condensates produced by different smoking-machine conditions, because some countries have begun to mandate cigarette testing with alternative smoking-machine conditions.
The use of experimental animal studies to predict cancer risk is more qualitative than quantitative (Stratton et al. 2001). Most animals used in laboratory studies with smoke are obligate nose breathers. Furthermore, Wynder and Hoffman (1967) reported that the respiratory systems of laboratory animals differ qualitatively and quantitatively from those of humans in surface area, in the development of mucous membranes, and in having an enhanced glandular system that increases the fluid in the nasal passages. Despite these limitations, animal studies provide information that is not available from in vitro systems because animal studies permit the use of an intact host system with a full complement of endocrine, hormone, and immune effects and hepatic and extrahepatic metabolism (Eaton and Klaassen 2001). Animal studies are often used to confirm positive findings or to resolve conflicting results from in vitro assays and to study organ-specific effects. Animal studies provide valuable data in terms of biologic plausibility, mechanisms of action, and causality. Animal studies of chronic diseases such as cancer can be less expensive than human clinical studies, and they also allow the use of invasive procedures (Devlin et al. 2005).
The smoke and smoke condensate animal bioassay literature is extensive and was reviewed by IARC in 2004. A synopsis follows of similar literature with a focus on studies made available since the publication of that review.
Dermal Application of Cigarette Smoke Condensate
Studies have used mouse skin as the test tissue in experiments carried out during the past 35 years, and the results from various laboratories have been similar with respect to the overall degree of carcinogenic activity of cigarette smoke condensate and the major differences in activity from cigarettes with different designs. Cigarette smoke condensate produces both benign and malignant tumors on mouse skin. The induced tumors are usually of epidermal origin. The carcinogenic potency of the cigarette smoke condensate depends on the tobacco variety, the composition of the cigarette paper, and the presence of additives. Subtle differences in smoking techniques, storage conditions for cigarette smoke condensate, and procedures for animal exposure do not appear to critically affect the results (IARC 2004). Researchers also conducted a limited number of skin-painting studies in other animal species including Syrian golden hamsters (Bernfeld and Homburger 1983) and rabbits (Graham et al. 1957).
In early skin-painting experiments with mice, researchers examined the tumorigenic activity of smoke condensates from reference cigarettes, from cigarettes made with different reconstituted tobacco sheets, or from mixtures of smoke condensates from reference cigarettes and reconstituted tobacco sheets made with 8-percent sodium nitrate as a tobacco additive (Dontenwill et al. 1972). Three preparations were tested: smoke condensate, dry smoke condensate without volatile smoke components, and condensate from vapor phase smoke. The smoke and the dry smoke condensates were equivalent in their ability to induce tumors, but the condensate from vapor phase smoke was nearly ineffective. The manufacturing process used to prepare the reconstituted tobacco sheet was a factor in the tumorigenic activity of the smoke condensate. Sodium nitrate reduced the tumorigenic activity of smoke condensate when added to the tobacco, to the reference cigarettes, or to the reconstituted tobacco sheet.
Subsequent studies continued to evaluate reference and experimental cigarettes constructed of tobacco-derived materials in dermal tumor promotion studies with female SENCAR mice (Meckley et al. 2004a,b). Cigarette smoke condensate from 1R4F reference cigarettes, which was applied to the skin of mice three times per week for 29 weeks, produced significant, dose-dependent increases in both the number of tumor-bearing animals and in the total number of tumors in mice treated first (initiated) with the carcinogen 7,12-dimethylbenz[a]anthracene (DMBA). The tumors were papillomas and squamous cell carcinomas; papillomas were still progressing toward carcinomas at the end of the study. Animals in the high-dose group demonstrated treatment-related damage to the treated dorsal skin. The damage was described as peeling skin, erythema, and sores. The effects on the dorsal skin occurred at a lower incidence in the middle-dose group. Dose-dependent histologic changes in nonneoplastic skin at the treatment site were characterized by increased epidermal thickness (acanthosis) and hyperkeratosis. Significant increases were reported in the ratios of organ to body weight for the kidneys, liver, and spleen and in organ weight and ratios of organ weight to brain weight for the liver and spleen in the mid- and high-dose groups compared with those for the control group, which was initiated with DMBA but not promoted with condensate. Histologic examinations revealed an increase in extramedullary hematopoiesis of the spleen in the high-dose group.
To increase the filling power of tobacco, manufacturers developed processes to impregnate shredded tobacco with volatile materials and then rapidly remove them to expand the cellular structure of the leaf, thereby reducing the density of the tobacco filler. The expanded tobacco was shown to have a high burn rate and irritating smoke (Browne 1990). The reduced cigarette weight, increased filling power, and increased burn rate reduced the number of puffs, which, in turn, reduced delivery of tar and nicotine (Abdallah 2003a). Expanded tobacco is included in commercial cigarettes, and the amount of expanded tobacco as a percentage of the tobacco mass increases from approximately 15 percent in full-flavored cigarettes to 50 percent in ultralight brands (Theophilus et al. 2004). Other scientists reported that concentrations of most chemicals measured in the smoke of cigarettes with puffed, expanded, or freeze-dried tobacco were significantly reduced compared with those in control cigarettes (Hoffmann et al. 2001).
In a study by Theophilus and colleagues (2003b), condensates from the smoke of cigarettes constructed with 100-percent tobacco expanded with dry ice or Freon-11 (trichlorofluoromethane) produced similar numbers of tumor-bearing animals and total tumors in DMBA- initiated mice. Animals in the group treated with a low dose of condensate from smoke of tobacco expanded with Freon had a significantly longer median time to onset of tumors and significantly more total tumors than animals in the group treated with a low dose of condensate from smoke of tobacco expanded with dry ice. No biologically significant nonneoplastic changes were observed in internal organs or treated dorsal skin. Smoke from the tobacco expanded with dry ice contained significantly higher concentrations of CO2, acetone, formaldehyde, catechol, nitric oxide, and NATB than did smoke from the Freon-expanded tobacco.
In other research, Theophilus and colleagues (2003a) studied smoke from cigarettes constructed with 100-percent propane-expanded tobacco. They found that the smoke had significantly higher concentrations of total particulate matter, nicotine, tar, CO, CO2, ammonia, catechol, hydroquinone, phenol, p- and m-cresol, nitric oxide, NATB, and NNK than did the smoke from Freon- expanded tobacco. No biologically significant nonneoplastic differences in internal organs or treated dorsal skin were observed among animals treated with condensate from cigarettes containing propane-expanded tobacco compared with animals treated with condensate from cigarettes containing Freon-expanded tobacco. Smoke condensates from cigarettes made with Freon-or propane-expanded tobacco produced similar numbers of tumor-bearing animals and total tumors in DMBA-initiated mice.
In another study, Theophilus and colleagues (2004) treated mice with smoke condensate from cigarettes constructed with increasing percentages of expanded shredded tobacco stems. In general, there was a pattern of increasing numbers of tumor-bearing animals and total tumors with increasing doses of tar among groups of mice treated with low, medium, or high concentrations of expanded shredded tobacco stems. This pattern was not present across these groups at a given tar level. The control group treated with condensate from cigarettes without expanded shredded tobacco stems showed a dose-dependent increase in the percentage of animals with tumors and in the total number of tumors compared with DMBA-initiated animals in the solvent (vehicle) control group not treated with smoke condensate. Cigarettes containing expanded shredded tobacco stems produced lower concentrations of some chemicals in mainstream smoke than did cigarettes that did not contain expanded shredded tobacco stems, but the concentrations were not consistently reduced in a dose-dependent manner.
In vivo and in vitro analyses support the hypothesis that short-term measures such as cytotoxicity, cellular proliferation (hyperplasia), generation of free radicals, and inflammation are involved in tumor promotion produced by cigarette smoke condensate (Curtin et al. 2004a). Other studies found that in addition to promoting tumors, cigarette smoke condensate and its fractions can act as tumor initiators, tumor accelerators, and cocarcinogens when applied together with other chemicals such as B[a]P and complete carcinogens (Wynder and Hoffmann 1961; Hoffmann and Wynder 1971; Hecht 2005).
The results from studies of dermal application of cigarette smoke condensate suggest a tissue-specific response to the chemicals in cigarette smoke that undergo covalent binding to DNA. Investigators have detected adducts in the skin, lung, heart, kidney, liver, and spleen of female ICR mice treated topically with cigarette smoke condensate from a commercial U.S.-blended unfiltered cigarette (Randerath et al. 1986, 1988; Reddy and Randerath 1990). In one study, dermal application of condensate from the smoke of 1R4F reference cigarettes three times per week for one or four weeks induced DNA adducts in the skin and lung tissue of male CD-1 mice (Lee et al. 1992). The relative adduct labeling values in skin were highest after one week of exposure and did not increase after four weeks. DNA adduct levels in the lung increased between one week and four weeks of treatment with condensate. Skin adducts declined to less than one-half the values of the first week by four weeks after cessation of exposure to condensate. In contrast, adduct levels in the lung continued to increase during the four weeks after cessation of exposure. Adduct levels increased with the total amount of tar applied weekly. The dose-response relationship was especially evident in lung tissue. In another study, treatments three times per week with similar concentrations of condensate from 1R4F cigarettes for 29 weeks resulted in an increase in DNA adducts in skin and dose- and time-dependent increases in DNA adducts in lung and heart tissues of female SENCAR mice (Brown et al. 1998).
Inhalation Studies with Cigarette Smoke
Historically, animals have not proven to be good models for the type of lung tumors induced by cigarette smoke in humans. Inhalation exposure to cigarette smoke leads to a reduction in the respiratory rate, and nontransgenic animals and animal strains with a low background incidence of lung tumors often do not develop an excess of lung tumors of any type. Researchers have attempted to induce lung cancer by exposure to cigarette smoke in several animal species, including rabbits, monkeys, dogs, and hamsters and other rodents. Hamsters developed laryngeal tumors but not tumors in the lower respiratory tract, and dogs developed epidermoid and bronchioloalveolar carcinomas (Coggins 2002; IARC 2004; Witschi 2005). Rodents tended to develop adenomas arising in the periphery of the lung rather than bronchial tumors arising centrally (Stratton et al. 2001). A study by Hutt and associates (2005) was the first to describe successful induction of lung tumors in mice after a lifetime whole-body exposure to mainstream cigarette smoke. Many animal studies used exposure chambers that permit whole-body exposure to cigarette smoke. Modern nose-only exposure tubes that allow body heat to dissipate are regarded by some as superior to whole-body exposure chambers because they eliminate dosing by nonrespiratory routes and allow the test concentration delivered to the animal to be closer to the concentration delivered to the system, by avoiding loss of the test compound on the walls, loss on the skin and fur of the animals, sedimentation and impaction of aerosol particles in the chamber, and chemical reactivity in the chamber (Pauluhn 2005). Table 3.1 contains data on lung tumor incidence from studies of carcinogenicity in rodents that used inhalation exposure to cigarette smoke.
Table 3.1
Selected chronic carcinogenicity studies in mice and rats with inhalation exposure to cigarette smoke.
Mouse. Witschi and colleagues (1997a) demonstrated that mouse lung tumors developed in the peripheral lung as areas of hyperplasia that progress to adenocarcinomas. In subsequent research, Witschi and colleagues (2002) studied male Balb/c and SWR mice exposed to a mixture of 89-percent sidestream smoke and 11-percent mainstream smoke from 1R4F reference cigarettes (Witschi et al. 2002). As reported in the previous studies (Witschi et al. 1997a,b), the investigators included a four-month recovery period to increase the development of lung tumors. In both strains, they observed increases in lung tumor multiplicities (average number of tumors per lung) (0.44 ± 0.13 and 0.35 ± 0.14, respectively) and lung tumor incidences (number of tumor-bearing mice per total number of treated mice) (33% in treated Balb/c mice versus 20% in controls and 19% of treated SWR mice versus 4% in controls, respectively) (Table 3.1) after exposure to cigarette smoke. Only the lung tumor multiplicity in treated SWR mice was statistically different from that in SWR controls exposed to air only. These investigators found that strain A/J mice were more susceptible to carcinogen-induced lung tumors than were Balb/c or SWR mice (Witschi et al. 2002). The same exposure regimen showed that in male strain A/J mice, the lung tumor multiplicity was significantly higher among the exposed mice than among the air-only controls, and there was a good correlation between exposure (average concentration of cigarette smoke multiplied by exposure duration) and lung tumor multiplicity (Witschi et al. 2002). Proliferative pulmonary lesions were categorized as focal alveolar epithelial hyperplasia, alveolobronchiolar adenomas, and alveolobronchiolar adenocarcinomas. Although it was possible to achieve a dose-related increase in lung tumor multiplicity in A/J mice with this exposure protocol, mice exposed to cigarette smoke had fewer adenomas with car-cinomatous foci or adenocarcinomas (malignant tumors) than did air-only controls (Witschi et al. 2002).
In a later study, Witschi and colleagues (2004) used a similar exposure regimen with five months of whole-body exposure to smoke from 2R4F reference cigarettes (89-percent sidestream and 11-percent mainstream smoke), followed by a four-month recovery period. This regimen produced a significant increase in lung tumor multiplicity and tumor incidence compared with the air-only controls although the response to the high dose was slightly less than to the medium dose (Table 3.1) in male strain A/J mice. The authors attribute the flat dose- response curve to the weak lung carcinogenicity of cigarette smoke in mice. The tumors were described as bronchioloalveolar adenomas.
Curtin and associates (2004b) studied effects of subchronic exposure to mainstream smoke from 1R4F reference cigarettes in male RasH2 transgenic mice, which carry the human C-HA-RAS oncogene, and A/J mice. Mice had whole-body exposure for 20 weeks or nose-only exposure for 28 weeks. Results indicated that whole-body exposure may be more effective than nose-only exposure for inducing statistically significant changes in tumor multiplicity and tumor incidence. One concentration of cigarette smoke was used in the whole-body experiments, and three concentrations were used in the nose-only experiments. Both exposure regimens included a 16-week recovery period. With whole-body exposure, microscopically confirmed tumor incidence and tumor multiplicity were significantly greater in the exposed animals than in the sham-exposed animals in both mouse strains.
Hutt and colleagues (2005) developed a model that achieved a 10-fold increase in hyperplastic lesions, a 4.6-fold increase in adenomas and papilloma, a 7.25-fold increase in adenocarcinomas, and a 5-fold increase in metastatic pulmonary adenocarcinomas in mice with lifetime whole-body exposure to cigarette smoke compared with lesions in air-only sham controls. The B6C3F1 strain of mice used in this study have low background incidence of lung tumors compared with that for A/J mice used in other studies. The female mice were exposed to mainstream smoke from an unfiltered 2R1 reference cigarette for 30 months. An increase in lung hyperplasia and neoplasia was first seen in mice exposed to TPM that died spontaneously between 540 and 720 days after the initial exposure. At the end of the study, the survival of mice exposed to smoke was significantly longer than that of the sham-exposed controls possibly because of reduced food consumption leading to lower body weight and a lower incidence and delayed onset of other types of cancers. Animals exposed to TPM also had a statistically significant increase in incidence of benign and malignant proliferative lesions in the nasal cavity. In contrast to other studies using a mouse model, this study achieved a significantly greater incidence of adenocarcinomas in treated animals (67 of 330, 20.3 percent) than in sham-exposed controls (9 of 326, 2.8 percent).
Rat. Female Fischer-344 (F-344) rats received daily nose-only exposure to the smoke of experimental cigarettes for 126 to 128 weeks (7 cigarettes per 8-hour day, 5 days per week) (Dalbey et al. 1980). The mean delivery of smoke particulate from 85-mm unfiltered cigarettes (National Cancer Institute, code 16) was 18.4 mg per cigarette. The exposure chamber consisted of holding tubes attached to the side of a 350-mL chamber containing a mixture of cigarette smoke and room air. The two control groups consisted of unexposed and sham-exposed controls. Survival in the smoke-exposed rats was similar to that of the two control groups combined. Animals in the group exposed to smoke had significantly more tumors of the respiratory tract than did the combined control groups (Table 3.1). Compared with controls, rats exposed to cigarette smoke had significantly fewer tumors in the hypophysis, hematopoietic and lymphoid system, uterus, and ovary. The number of adrenal tumors and oral tumors in treated animals increased, but the changes did not reach statistical significance. Animals exposed to smoke also had a significant increase in dermal tumors (subcutaneous sarcomas) near ulcers on the front feet from pushing against the holding tubes during exposure compared with animals in the control groups.
In the same study of lifetime exposure, researchers observed nonneoplastic tumors throughout the respiratory tract of animals exposed to smoke (Dalbey et al. 1980). These lesions included hyperplastic and metaplastic areas in the epithelium of the upper airways (nasal turbinates, larynx, and trachea) and areas of the lung with focal alveolitis (accumulations of pigmented macrophages, alveolar epithelial hyperplasia, and alveolar fibrosis).Lesions in control animals were much smaller and less severe. Researchers observed fibrosis and thickening of arterioles in the papillary muscle of the heart. No other organ systems showed evidence of smoke-related pathology.
One study also used chronic whole-body exposure in an attempt to achieve higher lung doses of cigarette smoke in F-344 rats (Mauderly et al. 2004). Low (100 mg TPM/cubic meter [m3]) and high (250 mg TPM/m3) concentrations of mainstream cigarette smoke were used for exposures up to 30 months. Cigarette smoke was produced from unfiltered 1R3 reference cigarettes machine puffed twice per minute using a 70-mL, two-second puff and then diluted with air cleaned by a high-efficiency particulate air filter. Exposure to cigarette smoke significantly increased the incidences of nonneoplastic and neoplastic, proliferative lung lesions in female rats. Trends with exposure for all neoplastic lung lesions were highly significant for female rats. No trend with exposure was significant for males. Time to first observation of hyper-plastic lesions was shortened by exposure among female but not male rats. Both benign and malignant neoplasms were observed earlier in high-exposure female rats than in low-exposure female rats. Hyperplastic responses consisted primarily of focal alveolar epithelial hyperplasia. Benign neoplasms were bronchioloalveolar adenomas, and malignant neoplasms were bronchioloalveolar carcinomas. Mean absolute weights of lungs in male and female rats exposed to high concentrations of smoke were significantly greater than those in animals in the control groups. Nonproliferative changes more common in animals in high-exposure groups than in low-exposure groups were ciliated cuboidal cell metaplasia and squamous metaplasia in alveolar ducts. There was no consistent difference by sex in development of proliferative nasal lesions, and the incidence of nasal cavity neoplasms increased significantly in both male and female rats exposed to high concentrations of smoke. Most of the nasal cavity tumors were epithelial in origin, and the benign epithelial tumors were adenomas. Histologic changes in the nasal cavity, such as squamous metaplasia of transitional and respiratory epithelium, mucous cell metaplasia and hyperplasia, and inflammatory infiltrates, were more common or more severe in the rats exposed to high concentrations of smoke.
Carcinogenicity bioassays should be conducted for a major portion of the test animal's lifetime. Short-term (subchronic) exposure studies are primarily performed to provide information on target organs of repeated exposure. Short-term, nose-only exposures to mainstream smoke produced treatment-related histopathologic changes in the respiratory tract and in clinical chemistry parameters in male and female Sprague-Dawley rats (Coggins et al. 1989; Ayres et al. 2001; Terpstra et al. 2003). Animals exposed to cigarette smoke had significantly more chronic active inflammation, epithelial hyperplasia, atrophy of the olfactory epithelium, and squamous metaplasia of the nasal passages and larynx. Other histopathologic changes included increased counts of intra-alveolar brown-gold macrophages and bronchial goblet cells. There was a dose-dependent trend toward increased severity of the effects with increased exposure. Some of the effects such as laryngeal squamous metaplasia were not completely reversed during a recovery period.
A U.S.-tobacco-blend cigarette containing the additive 1-menthol and other conventional processing aids and flavoring ingredients was compared with a reference cigarette comprised of a similar tobacco blend in a 13-week inhalation study of toxicity in male and female F-344 rats (Gaworski et al. 1997). Only one concentration of 1-menthol (5,000 ppm) was used. Three dose levels of cigarette smoke were tested for each cigarette. Both cigarette varieties produced similar dose-related histologic changes in the respiratory tract and increases in ratios of lung weight to body weight. The researchers noted that although lesions in the trachea and larynx related to cigarette smoke were similar in incidence, the degree of the response was slightly more severe in some groups of female rats exposed to mentholated cigarette smoke than it was in those exposed to nonmentholated cigarette smoke.
Theophilus and colleagues (2003a,b, 2004) performed several 13-week nose-only inhalation studies with Sprague-Dawley rats to evaluate the toxic effects of expanded materials derived from tobacco (Theophilus 2003a,b, 2004). The exposure regimen consisted of one hour of exposure per day, five days per week, for 13 weeks, followed by a 13-week recovery period. Male and female rats were exposed to mainstream smoke from cigarettes constructed of 100-percent tobacco expanded with dry ice or Freon-11 (Theophilus et al. 2003b) or tobacco expanded with propane or Freon-11 (Theophilus et al. 2003a). Animals exposed to cigarette smoke demonstrated chronic active inflammation and epithelial hyperplasia of nasal tissues and ventral squamous metaplasia of the larynx that appeared to increase in severity with increasing doses. Treated animals also had significantly more non-pigmented macrophages and brown-gold macrophages and evidence of chronic active inflammation of the larynx than did air-only controls. Most of the histologic changes resolved after a 13-week recovery period (Theophilus et al. 2003a,b). A separate study was conducted to evaluate the toxic effects of different percentages of expanded shredded tobacco stems (Theophilus et al. 2004). Overall, exposure to mainstream smoke from cigarettes constructed of 9.25-percent, 18.5-percent, or 25-percent expanded shredded stems failed to produce signs of increased or decreased toxicity relative to the control cigarettes that did not contain expanded shredded stems. At the highest dose, animals in all the groups exposed to expanded shredded stems had significant increases in the severity of nonpigmented macrophages in left and apical regions of the lung compared with those in unexposed animals. Treatment groups with the medium (18.5 percent) and high (25 percent) content of expanded shredded stems also had a significant increase in the severity of nonpigmented macrophages and goblet cells in the right diaphragmatic region of the lung at the highest dose compared with animals treated with smoke from control cigarettes containing zero-percent expanded shredded stems. Theophilus and colleagues did not describe the composition or tobacco blend in the control cigarettes or in the cigarettes made with expanded shredded stems. They stated that the tobacco blend and cigarette configuration were comparable between test and control cigarettes and that the main difference was the percentage of expanded shredded stems in the test cigarettes.
Cardiovascular and Cerebrovascular Studies in Animals
Some animal models show promise for studying the development of cardiovascular disease induced by cigarette smoke. For example, researchers have proposed an elastase-perfusion mouse model for aortic aneurysms induced by cigarette smoke (Buckley et al. 2004). Another example is the cockerel as a model for arteriosclerosis (Penn et al. 1983, 1992, 1996; Penn and Snyder 1988). Cockerels are sensitive to the plaque-promoting effects of chemicals administered by inhalation or injection. Inhalation of mainstream cigarette smoke or the vapor phase smoke component, 1,3-butadiene, was shown to promote plaque development in cockerels, but CO or an injection of NNK or solubilized concentrated cigarette smoke condensate from an unventilated 2R1 reference cigarette did not promote plaque development in cockerels. At high doses, intramuscular injections of PAH compounds with different carcinogenic potencies also led to arteriosclerotic plaque formation. Sidestream smoke was more effective than mainstream smoke in stimulating aortic plaque development in the cockerel model.
Investigators reported that tissue injury induced by oxidative stress, altered serum lipids, increased blood pressure, and endothelial damage were other possible factors in cardiovascular injury from cigarette smoking (Stratton et al. 2001). In another study, inhalation exposure to cigarette smoke produced evidence of oxidative stress in the hearts of Balb/c mice (Koul et al. 2003). After 10 weeks of whole-body exposure for 60 minutes per day to the smoke from five commercial filter-tipped cigarettes, mice had significantly lower concentrations of glutathione and higher concentrations of lipid peroxides, glutathione peroxidase, glutathione reductase, and catalase than did unexposed mice. Serum triglycerides, total cholesterol, LDLc, and the ratio of total cholesterol to HDLc were also significantly higher, and HDL and the ratio of HDLc to LDLc were significantly lower in the group exposed to smoke. Concomitant administration of α-tocopherol prevented some of the smoke-induced changes.
In one study, whole-body exposure to smoke from a 2R1 reference cigarette three times per day for 30 days resulted in a significant increase in the formation of 8-oxo-dG, a marker of oxidative damage, in the heart tissue of male Sprague-Dawley rats, compared with the concentration in unexposed controls (Park et al. 1998). Cigarettes were smoked for 15 to 20 minutes to a fixed butt length in a 500-mL flask with air pumped into the flask. The reduced glutathione content and the oxidative state of glutathione in heart tissue were not significantly different from those in controls. In another study, whole-body exposure to low concentrations of cigarette smoke resulted in impaired oxidative function in cardiac mitochondrial cells; increased intracellular, low-molecular- weight iron that can play a role in redox reactions; and reduced α-tocopherol during cardiac ischemia and reperfusion in female Sprague-Dawley rats (van Jaarsveld et al. 1992). These findings suggested a mechanism involving oxidant radicals. Twice a day for two months, smoke was introduced by inserting a lit cigarette into a hole in the exposure chamber and allowing smoke to be drawn into the chamber for 5 seconds, followed by room air for 55 seconds. This procedure was repeated until the cigarette extinguished (approximately 10 minutes). Carboxyhemoglobin concentrations in rats exposed to smoke or air were not statistically different.
Scientists reported that hepatic uptake of chylomicrons was significantly lower in Sprague-Dawley rats with whole-body exposure to the smoke of two unfiltered, king-size, GPC-brand cigarettes (35 to 40 mL per puff, 20 puffs per cigarette) than was uptake in sham-exposed controls (Pan et al. 1993). Animals were exposed to the smoke for 10 minutes, four times per day, for 10 days. In addition, more chylomicrons remained in the hearts of rats exposed to smoke than in the hearts of controls. Hepatic uptake and residence time in heart tissues also changed when chylomicrons were administered to unexposed animals that had been previously exposed to smoke. Smoke exposure increased the thiobarbituric acid reactive substance measurement in chylomicrons, a measure of lipid peroxidation. In a subsequent series of experiments, whole-body exposure to the smoke of two 2R1 reference cigarettes (35 to 40 mL per puff, 20 puffs per cigarette) for 10 minutes, six times per day, for 10 days, significantly increased postprandial serum triglyceride and chylomicron concentrations, decreased hepatic uptake of chylomicron remnants, and increased plasma postheparin lipoprotein lipase activity. Hepatic lipase activity was similar in rats exposed to smoke and controls (Pan et al. 1997). In another study, subchronic (14 or 90 days) but not acute (1 day) whole-body inhalation exposure to cigarette smoke resulted in significantly increased cholesterol, triglyceride, and phospholipid levels in the serum, hearts, aortas, and lungs of male Sprague-Dawley rats (Latha et al. 1988). Changes in serum lipoproteins included decreases in HDLc, triglycerides, and phospholipids and increases in LDL and very-low-density lipoprotein cholesterol, triglycerides, and phospholipids. Other alterations in lipid metabolism included increased hydroxymethylglutaryl- CoA reductase activity, decreased lipoprotein lipase activity in heart tissue, and increased lipoprotein lipase in adipose tissue.
Research in male hypercholesterolemic ApoE *−/*− mice suggested that five weeks of exposure to 1R4F cigarette smoke led to an increase in oxidized LDL immunoglobulin M (IgM) and antiphosphorylcholine IgM antibodies and a decrease in oxidized LDL IgG and lymphotoxin-β messenger ribonucleic acid (mRNA) expression in the spleen (Tani et al. 2004). Both IgM changes were associated with an increase in thickness of arterial intima. Animals were acclimated to cigarette smoke produced by a vacuum pump until smoke from one cigarette per day was tolerated. Mice exposed to cigarette smoke had significantly higher serum carboxyhemoglobin concentrations than those of air-only controls.
Researchers examined the aortic endothelium from male Sprague-Dawley rats by scanning and transmission electron microscopy after inhalation exposure to the smoke of two medium-tar cigarettes (19 mg of tar and 1.5 mg of nicotine) (Pittilo et al. 1982, 1990). Smoking-machine conditions were not provided in the description of study methods. Animals were anesthetized before exposure. The exposure was repeated 5 days per week during a 25-day period. Compared with sham-exposed controls, the rats exposed to smoke demonstrated marked morphologic evidence of endothelial damage that included increased blebs, microvillus-like projections, plasmalemmal vesicles, and Weibel-Palade bodies that store von Willebrand factor protein. No endothelial abnormalities were seen in rats that received nicotine by subcutaneous injections or by continuous subcutaneous pumps. These observations suggest that components of cigarette smoke other than nicotine are responsible for the endothelial cell changes associated with smoking. Researchers reported that male Sprague-Dawley rats with exposure to the smoke of five low-nicotine (1 mg) cigarettes for 20 to 30 minutes per day for four to six weeks had significantly higher mean arterial blood pressure after bilateral occlusion of the common carotid artery than did the sham-exposed controls (Bennett and Richardson 1990). Additionally, the time required to reach a maximum mean arterial blood pressure after occlusion was significantly less in the animals exposed to cigarette smoke versus the sham-exposed controls. Only one smoke concentration was used in this study. Using anesthetized, mechanically ventilated rats as an in vivo model, researchers showed that cigarette smoke produced a biphasic change in the diameter of the cerebral arterioles and an increase in mean arterial blood pressure in rats (Iida et al. 1998). An initial vasoconstriction was seen in animals breathing the smoke but not in animals receiving an intravenous infusion of nicotine. These findings led researchers to conclude that a smoke constituent other than nicotine was responsible for the early vascular change. Thromboxane A2 was pro-posed as the agent responsible for the vasoconstriction. Others have determined that cigarette smoking, but not the use of transdermal nicotine or smokeless tobacco, increased concentrations of thromboxane A2 (Wennmalm et al. 1991; Benowitz et al. 1993).
Cardiovascular changes were observed in several studies of short-term and lifetime inhalation cigarette smoke exposure in rodents. Investigators studied male Wistar rats with whole-body exposure to the smoke of an unidentified commercial cigarette for 30 days. They observed a significant increase in left ventricular systolic diameter and a significant reduction in systolic shortening fraction and ejection fraction compared with those in unexposed controls (de Paiva et al. 2003). No change in heart rate or heart weight was seen under the exposure conditions of this study. Another study demonstrated a significant increase in heart weight in female, but not male, Sprague-Dawley rats with 13 weeks of daily nose-only inhalation exposure to the smoke of a custom-blended experimental cigarette smoked under FTC conditions (Coggins et al. 1989). Other investigators conducted a 13-week inhalation study of male and female F-344 rats exposed to smoke of mentholated or nonmentholated cigarettes. Male rats exposed to medium or high doses and female rats exposed to high doses of smoke from mentholated cigarettes, machine smoked under FTC conditions, developed a significant increase in the ratio of heart weight to body weight (Gaworski et al. 1997). Male and female rats exposed to high doses of smoke from nonmentholated cigarettes also had a significant increase in cardiomegaly (high ratio of heart weight to body weight). The difference between treated and control animals was no longer significant after a six-week recovery period. Dalbey and colleagues (1980) observed fibrosis and thickening of arterioles in the heart papillary muscle of female F-344 rats with daily nose-only inhalation exposure for 126 to 128 weeks to smoke from unfiltered experimental cigarettes. No smoke-related pathologic changes to the large vessels were detected from the one concentration of smoke (10 percent) that was used.
Studies using dermal applications of smoke condensate or inhalation exposure to cigarette smoke demonstrated that chemicals in cigarette smoke underwent covalent binding to heart tissue DNA in laboratory animals (Randerath et al. 1986, 1988; Reddy and Randerath 1990; Brown et al. 1998; Gupta et al. 1999). Studies of cigarette smokers showed that the heart tissue contained more DNA adducts than that from nonsmokers or former smokers (Van Schooten et al. 1998). They also demonstrated a linear relationship between DNA adduct levels and daily cigarette smoking. Furthermore, higher DNA adduct levels were associated with a higher degree of coronary artery disease.
Respiratory Studies in Animals
Exposure to chemicals in cigarette smoke affects the function of the respiratory system in laboratory animals and humans. Notably, exposure to cigarette smoke affected airway mucociliary function (Shephard 1978; Wanner 1985; Finch et al. 1995). Another researcher demonstrated that exposure resulted in a dose-dependent inhibition of lung clearance and increased absorption of components of the inhaled smoke through the tracheo-bronchial airways, especially where particle deposition occurred and mucociliary clearance was less efficient, specifically at the ridges within bifurcations and in posterior sections of tubular airways (Martonen 1992). Studies have shown that the activity of xenobiotic metabolizing (cytochrome P-450) enzymes in human lung tissue is likely sufficient to cause in situ activation of pulmonary toxicants (Castell et al. 2005). Species differences in enzyme activities have led some to question the use of animal data to predict toxic effects in humans from chemicals requiring bioactivation to reactive metabolites (Castell et al. 2005). Short-term assays to evaluate the components of cigarette smoke that impair mucociliary function were described in the "Cytotoxicity" section earlier in this chapter.
Persistent pulmonary inflammation from repeated exposure to cigarette smoke may lead to more severe alterations in the structure and function of the lung (Stratton et al. 2001). For example, researchers concluded that emphysema in cigarette smokers reflects a low-level, chronic inflammatory process in the lower respiratory tract with an imbalance of protease and antiprotease activities leading to the degradation of connective tissue (Churg et al. 2002).
Syrian golden hamsters have been used extensively to study the pathogenesis of emphysema. They show a pattern of inflammatory airway response and impaired activity of antioxidants (superoxide dismutase and catalase)—a pattern similar to that in humans with repeated exposure to cigarette smoke (Hoidal and Niewoehner 1982; McCusker and Hoidal 1990). Rat strains were shown to be more resistant to the induction of emphysema by exposure to cigarette smoke, but susceptibility in mice was strain specific (Groneberg and Chung 2004). Research on emphysema induced by cigarette smoke in animals has not consistently demonstrated progression of the disease (March et al. 1999). In a comparative study of B6C3F1 mice and F-344 rats, the mouse strain displayed more morphometric changes (parenchymal air-space enlargement, volume density of alveolar air space, and loss of alveolar tissue) and significantly more neutrophils within inflammatory lesions in the lung. Morphometric differences in the mice at 13 months were greater than those at 7 months. This finding suggests that in mice, emphysema induced by cigarette smoke may be progressive. Animals received a whole-body exposure to the smoke of 2R1 reference cigarettes that were machine smoked (two 70-mL puffs per minute) and diluted with filtered air to achieve a chamber concentration of 250 mg of TPM/m3. The exposure duration was six hours per day, five days per week. The investigators concluded that the type of inflammatory response may be a determining factor for differences in susceptibility to emphysema induction by cigarette smoke among test species.
Animal models can readily be used to detect and quantitate the pulmonary inflammatory response to inhaled compounds or mixtures, and the literature in this area was reviewed (Stratton et al. 2001; IARC 2004). An analysis of bronchoalveolar lavage (BAL) fluid for cellular and biochemical indicators of inflammation allows quantitation of the pulmonary inflammatory response of rodents to inhaled cigarette smoke (Churg et al. 2002; Shapiro et al. 2003). The differential cell count and the functioning of cells obtained by the BAL technique can be used to classify the type of inflammatory response, and the biochemical content of the BAL fluid can be used to detect release of various cytokines and alterations in pulmonary surfactant (Stratton et al. 2001; Miller et al. 2002).
Response of inflammatory cells, cytokine profiles, enlargement of air space, and mechanical properties of the lung (elastance) differed among mouse strains after exposure to cigarette smoke (Guerassimov et al. 2004). In one study, emphysema-resistant (ICR) and emphysema-sensitive (C57BL/6) mouse strains showed differences in BAL cytokine and chemokine responses following a nose-only inhalation exposure to 2R1 reference cigarette mainstream smoke (two-second, 35-mL puff, once per minute) for two hours per day for seven days (Obot et al. 2004). Test concentrations were achieved by diluting mainstream smoke with fresh, conditioned air. There was a significant dose response for chemokines and cytokines (KC, JE, MIP-1α, MIP-2, RANTES, interleukin (IL)-17, SDF-1β) that recruit or activate neutrophils and other cell types in ICR mice, and a significant dose-response change in thymus- and activation-regulated chemokines was noted in C57BL/6 mice. Other researchers found that in contrast to emphysema-resistant ICR mice, emphysema-sensitive mouse strains (DBA/2 and C57BL/6J) showed a decrease in BAL antioxidant capacity after acute whole-body exposure to smoke (five cigarettes in 20 minutes) from a commercial, Virginia-tobacco-type cigarette (Cavarra et al. 2001). The animals that had lifetime exposure to the smoke (three cigarettes per day for 90 minutes, five days per week, for seven months) had decreased lung elastin content and developed emphysema. In a study of male ICR mice exposed five days per week for two weeks to mainstream smoke from commercial, unfiltered, high-tar cigarettes (1-second puff of 20-mL volume at 10-second intervals and 45 puffs per cigarette), the lungs showed evidence of senescence of alveolar epithelial cells (increased β-galactosidase activity, lipofuscin accumulation, and P21CIP1/WAF1/SDI1 protein in type II cells) (Tsuji et al. 2004). The researchers proposed that the senescence prohibited lung epithelial cells from proliferating and repopulating epithelial cells lost to apoptosis during emphysema.
Bartalesi and associates (2005) also studied whole-body exposure to cigarette smoke from three commercial, filter-tipped, Virginia-tobacco cigarettes per day, five days per week, for up to 10 months. This exposure produced epithelial cell injury, loss of cilia in the airways, and a positive reaction for mouse neutrophil elastase. The findings suggested degradation of lung elastin in emphysema-sensitive (C57BL/6J and DBA/2) mouse strains. The C57 strain of mice is moderately deficient in serum α 1-proteinase. Overt emphysema in the C57 strain was characterized by disseminated foci of severe emphysema interspersed by normal parenchyma. In DBA/2 mice, the foci of emphysema were scattered in a network of uniformly dilated parenchyma. Other differences were a greater fibrotic reaction and faster development of emphysema in DBA/2 mice (three months versus six months in C57 mice), and more extensive goblet cell metaplasia and immunohistochemical reaction for IL-4, IL-13, and MUC5AC (a secreted mucin) in C57 mice.
Further research with genetically modified mice explored the role of α1-antitrypsin (AAT), elastases, and tumor necrosis factor-alpha (TNFα) in emphysema induced by cigarette smoke. In one study, mice deficient in AAT (C57BL/6J *pa/*pa [pallid]) developed diffuse, panlobular emphysema affecting the entire air space, and C57 mice with normal concentrations of AAT developed more localized centrilobular emphysema (Takubo et al. 2002). Other more evident changes in the pallid mice with low concentrations of AAT after daily sub-chronic exposure (six months) to 2R1 cigarette smoke were increased T-cell inflammation in the alveolar wall and a reduced ability of the lungs to distend under pressure (compliance). Other investigators studied mice deficient in NE*−/*− or MMP-12*−/*−. The animals failed to develop air-space enlargement after six months of exposure to cigarette smoke from an unfiltered reference cigarette (Hautamaki et al. 1997; Shapiro et al. 2003). The investigators concluded that neutrophil elastase is required for recruitment of neutrophils and macrophages and for activation of MMP, which solubilizes extracellular matrix proteins including elastin. Other investigators reported that mice deficient in MMP (MMP*−/*−) that had a single exposure to the whole smoke of four 2R1 reference cigarettes did not show the same early elevations in lavage neutrophils, desmosine, or hydroxyproline that are seen in mice with normal levels of MMP activity (MMP*+/*+) (Churg et al. 2002). In a later study, these investigators reported that the absence of TNFα receptors is protective against infiltration of inflammatory cells, breakdown of lung matrix, and air-space enlargement in mice lacking P55 and P75 TNFα receptors (TNFRKO mice) after exposure to whole smoke from four 2R1 reference cigarettes five days per week for six months (Churg et al. 2004).
Several studies have shown that subchronic and chronic exposure to cigarette smoke produced evidence of respiratory tract toxicity that leads to emphysema in rats. In one study, the total glutathione, reduced glutathione, and protein-bound glutathione content in lung tissue of Sprague-Dawley rats with whole-body exposure to smoke from 2R1 reference cigarettes for 30 days, three times per day, were significantly lower than those in unexposed controls (Park et al. 1998). Oxidized glutathione increased significantly in rats exposed to smoke. Smoke exposure also produced a treatment-related increase in 8-oxo-dG DNA levels in the lungs. Cigarettes were smoked for 15 to 20 minutes to a fixed butt length in a 500-mL flask with air pumped into the flask.
Researchers found increased lung IL-4 and MMP-12 levels and decreased interferon-γ levels in Wistar rats after daily whole-body exposure to the smoke of 20 commercial unfiltered cigarettes six hours per day, five days per week, for three and one-half months (Xu et al. 2004). The changes were accompanied by pathologic evidence of emphysema in the form of inflammation, damage to airway epithelium and cilia, reduced mean alveolar number, air-space enlargement, and changes in pulmonary function.
Chronic nose-only exposure of female Sprague-Dawley rats to smoke from 2R1 reference cigarettes twice per day significantly reduced the disaturated phosphatidylcholine and surfactant protein levels in BAL fluid, but not in lung tissues, and significantly increased the albumin content of BAL fluid (Subramaniam et al. 1995). The researchers also observed increased surface compressibility and decreased respreading on expansion (respreadability index) of organic extracts of the BAL fluid from treated rats compared with those for room controls and sham-treated controls. Total levels of total lung phospholipids were not significantly different among the groups.
In selective reviews of the literature, Coggins (1998, 2002) summarized other nonneoplastic histopathologic changes observed in animals exposed to cigarette smoke:
-
pulmonary fibrosis in C57 mice accompanied by accumulation of lymphocytes and macrophages in the peribronchiolar and perivascular regions;
-
alveolar fibrosis, alveolitis, and bronchiolitis with accumulation of macrophages in F-344 rats;
-
granulomas in alveolar spaces and adjacent interstitial areas of all lobes of the lung in F-344 rats;
-
perivascular or peribronchiolar accumulation of lymphoreticular cells, fibrosis and cellular enlargement of peribronchiolar septa, hyperplasia of type II cells and septal fibrosis, and air-space enlargement in F-344 rats;
-
pulmonary edema, bronchial pneumonia, pulmonary fibrosis, emphysema, and cor pulmonale in beagle dogs that had tracheotomy; and
-
pleural thickening, alveolar fibrosis, and subpleural inflammation in beagle dogs without tracheotomy.
Reproductive and Developmental Studies in Animals
Fertility and Conception
Animal studies have suggested altered gonadotropin release, decreased luteinizing hormone surge, inhibition of prolactin release, altered tubal motility, and motility and impairment of blastocyst formation and implantation as possible mechanisms of fertility impairment among smokers (Stratton et al. 2001). In one study, male and female Sprague-Dawley rats received nose-only exposure to the smoke of 1R4F reference cigarettes (two-second puff, one puff per minute, 35-mL puff) for two hours per day, seven days per week, for four weeks before and during mating for males, and for two weeks before mating, during mating, and through gestation day 20 for females (Carmines et al. 2003). The investigators observed a statistically significant decrease in weight of seminal vesicles for males exposed to a low concentration or a medium concentration of smoke. Weight gains during pregnancy and mean uterine weight were significantly reduced in the female rats exposed to a high concentration of smoke. Fertility and conception endpoints unaffected by exposure to smoke were sperm count, motility, and morphology in males and corpora lutea, resorptions, implantation sites, and mortality in females. In another study, three months of whole-body inhalation exposure to mainstream smoke for two hours a day from a commercial, filter-tipped, high-tar cigarette mechanically smoked with a 50-mL syringe did not lead to a reduction in uterine weight or estrous cycle but did result in decreased estradiol concentration in rat uterine tissue compared with that in uterine tissues of sham-exposed control rats (Berstein et al 1999). The proliferation index and proportion of uterine tissue cells in S and G2/M phases were increased at three weeks of exposure for two hours per day. By three months, the differences in values from those of controls were no longer statistically different, but they were significantly lower than at three weeks, which the investigators attributed to a decline in the intensity of cell division.
In another study, Wistar rats received whole-body exposure to the smoke of a commercial cigarette from conception until parturition. Rats were exposed to cigarette smoke six hours per day, five days per week, for 11 weeks: 6 weeks before mating, 2 weeks during mating, and 3 weeks during pregnancy (Florek and Marszalek 1999). Three concentrations of cigarette smoke were monitored by CO concentration, and exposure was assessed by the determination of carboxyhemoglobin. Offspring were mated to produce two subsequent generations. The researchers observed an apparent dose-dependent reduction in the mating index, fertility index, and the number of pregnant rats, but no influence on the duration of pregnancy. This exposure regimen also resulted in a dose- dependent decrease in the mean number of animals rearing pups on the 21st postnatal day (Florek et al. 1999).
Researchers reported that the transport of preim-plantation embryos through the oviduct was retarded in golden hamsters with nose-only exposure to mainstream or sidestream smoke of unfiltered 2R1 reference cigarettes (DiCarlantonio and Talbot 1999). Low, medium, and high doses were produced by generating smoke from two, four, or six cigarettes. They observed that doses used in the study produced serum concentrations of cotinine within the range of those in women actively or involuntarily exposed to cigarette smoke during pregnancy. Animals were exposed to cigarette smoke (one puff per minute, 35-mL puff) 7 days per week, beginning 14 days before mating and continuing through day 3 of pregnancy. In females exposed to mainstream smoke, the increased percentage of embryos recovered from the oviducts on day three of pregnancy was dose dependent. The difference in these percentages for the hamsters in the medium-and high-dose groups and the control hamsters, who breathed only air, was statistically significant. The number of embryos retained in the oviducts of hamsters in all three groups exposed to sidestream smoke was significantly different from that for controls, but the researchers did not observe a dose-dependent pattern. The contraction rate of the oviductal muscle also decreased significantly during a single exposure to either mainstream or sidestream smoke and did not return to initial values during a 25-minute recovery period.
Researchers have evaluated the effects of individual components of cigarette smoke on reproduction in hamster oviducts in vitro. Many components act in a dose-response manner and inhibit oviduct function at concentrations found in cigarette smoke. Talbot and colleagues (1998) showed that cyanide concentrations in 2R1 cigarette smoke were sufficient to inhibit the ciliary beat frequency and time needed for an oocyte cumulus complex to travel through the oviduct to the ostium (oocyte cumulus pickup rate) in golden hamsters. Other constituents of cigarette smoke (acrolein, formaldehyde, phenol, and acetaldehyde) produced these alterations at concentrations that were 3 to 50 times higher than the corresponding concentrations in the smoke of an experimental 2R1 reference cigarette that was machine smoked under a single set of conditions (two-second puff, one puff per minute, 40-mL puff). All chemicals acted in a dose- dependent manner, and inhibition of the ciliary beat frequency for all except acrolein was at least partially reversible. The beat frequency of cilia treated with acrolein continued to decrease after the chemical was flushed out of the perfusion chamber. Tested individually, indole, 5-methylindole, quinoline, isoquinoline, hydroquinone, and substituted phenols (compounds present in the mainstream smoke of cigarettes), at picomolar to micromolar concentrations, inhibited oviductal functioning (ciliary beat frequency, oocyte pickup rate, and the contraction rate of infundibular smooth muscle) of golden hamster oviduct explants. Substitution of an ethyl or methyl group greatly increased the potency of the phenolic derivatives over that of the parent compound (Riveles et al. 2005). A recent study compared follicle loss and markers of apoptosis in the ovaries of mice exposed to mainstream cigarette smoke or B[a]P (Tuttle et al. 2009). Female mice received a nose-only exposure to mainstream smoke for eight weeks at a level equal to a pack-a-day habit in humans. Compared with mice exposed only to air, smoke-exposed mice had a significant reduction in the number of follicles. There was no increase in apoptotic follicles or other markers of cell death in response to cigarette smoke exposure. In vitro treatment of cultured ovaries with B[a]P did not increase apoptosis. The authors concluded that smoke exposure selectively reduced follicles in the primordial and transitional stages but that the loss was not due to apoptosis (Tuttle et al. 2009).
Fetal Effects
Researchers have demonstrated fetotoxicity from cigarette smoke exposure by reporting reduced fetal weight in rats and mice exposed during gestation. Reduced fetal weight is one of the most reproducible treatment-related effects. In utero exposure of fetal Sprague-Dawley rats to smoke from a king-size, filter-tipped, commercial cigarette on days 1 through 20 of gestation produced a significant reduction in fetal weight (Leichter 1989). For more than two hours, the adult female rats were exposed to the smoke of 10 lit cigarettes with the burning end of the cigarette placed inside a whole-body-exposure chamber. Litter size and placental weights were not different between rats exposed to smoke and pair-fed controls given the amount of food equal to that consumed by the smoke-exposed group. The increase in resorptions of implanted embryos in the group exposed to smoke was not significantly different from that in the controls. In a study of mainstream smoke from 1R4F cigarettes (two-second puff, one puff per minute, 35-mL puff), male Sprague-Dawley rats had nose-only exposure for four weeks before and during mating and female rats had nose-only exposure for two weeks before mating, during mating, and through gestation day 20 (Carmines et al. 2003). Researchers identified a significant decrease in mean fetal weight compared with that of the sham-exposed controls. The number of live and dead fetuses was unaffected by smoke exposure. A series of experiments with smoke from research cigarettes that varied in levels of nicotine, condensate, and CO demonstrated that the weight and length of fetuses from Sprague- Dawley rats was dependent on the intensity and duration of smoke inhalation (Reznik and Marquard 1980). All cigarettes were machine smoked with one set of conditions (two-second puff, one puff per minute, 35-mL puff). The number of exposures per day, the duration of the exposure in days, the number of puffs per cigarette, and the volume of air used to dilute the smoke were varied to create different exposure groups. The mean fetal weight and length decreased with increasing smoke concentrations, and fetuses of rats exposed to cigarette smoke two, three, or four times a day had significant reductions in weights and lengths compared with the fetuses of rats exposed for one period per day. Growth retardation was more extensive when smoke exposure occurred during the second half of pregnancy, but the reduction was less severe in the fetuses of rats exposed during the entire pregnancy. These effects could not be attributed to the CO concentration in the smoke alone, because the effects were more pronounced with exposure to the whole smoke than with exposure to the gas phase. The number of resorbed fetuses was not influenced by smoke exposure.
In one study, mice with the autosomal recessive curly-tail gene received nose-only exposure to the mainstream smoke of a commercial low-tar or high-tar cigarette for 20 minutes, once a day from day zero to day eight of pregnancy (Seller et al. 1992). Both cigarette varieties were smoked under the same smoking-machine conditions (two-second, 35-mL puff). The scientists observed similar levels of increased embryonic loss and retardation in embryonic development. The decrease in the mean somite number in the treated animals compared with that in the sham-exposed mice was statistically significant. Longer exposures (day 0 through day 17 of pregnancy) to smoke from the low-tar cigarettes resulted in a fivefold increase in intrauterine embryonic deaths, and live embryos weighed significantly less than those from the sham-treated group. Differences between the groups exposed to smoke from the high- or low-tar cigarettes were evident when exposure (10 minutes, three times a day) was restricted to days six, seven, and eight of pregnancy. The scientists reported that differences between the high-tar and low-tar treatment groups disappeared when the dose of the smoke from the low-tar cigarettes was increased. Weight loss in the treated pregnant mice was statistically significant, and weight loss was not dose dependent. Findings indicated a dose-response trend in the various dosing regimens, and the effect on embryonic survival and growth rate from exposure to the smoke of six cigarettes was greater than that of two cigarettes.
Curly-tail and C57BL strain mice received nose-only exposure on days six, seven, and eight of pregnancy to the smoke of a commercial low-tar or high-tar cigarette (Bnait and Seller 1995). One set of smoking-machine conditions (two-second puff, one puff per minute, 35-mL puff) was used to generate smoke from the low-tar and high-tar cigarettes, which was puffed over the noses of the test animals in individual chambers. Mice in both treatment groups were sacrificed on day nine. The embryos were removed, and embryonic cells from the fetal plate, surface ectoderm, pericardium, and heart were examined by scanning and transmission electron microscopy. In both strains, the morphology of the exterior of the neural cells, the surface ectoderm, the pericardium, and the heart were the same. Cells from embryos of females in the high-tar exposure group showed evidence of depressed metabolic activity, suggesting anoxic damage that persisted 20 hours after the exposure had ceased. In embryos from the low-tar group, changes were also present but were less marked than in embryos of mice in the high-tar group. No change occurred in the total cell number or in the number of dead cells or alteration in the mitotic index with either type of cigarette, but C57BL embryos of mice in the low-tar group had a significant reduction in the mitotic index compared with embryos of sham-treated controls.
Developmental Effects
Animal studies have suggested that even brief exposures from maternal smoking are detrimental to the very early embryo (Bassi et al. 1984; Collins et al. 1985; Lich-tenbeld and Vidíc 1989; Moessinger 1989; Seller and Bnait 1995). Prenatal exposure to cigarette smoke resulted in impaired growth and maturation of fetal lung, including reduced lung volume, lower internal surface area, fewer and larger alveoli, decreased lung interstitium and parenchymal elastic tissue, increased density of parenchymal interstitium, and apparent reduction in synthesis of surfactant.
Investigators in one study reported that a single four-hour, whole-body exposure to smoke from filter-tipped or unfiltered cigarettes (one puff per minute, 35-mL puff) and a single intranasal administration of cigarette smoke condensate induced DNA deletions in fetal C57BL/6J mice homozygous for the pink-eyed unstable mutation (Jalili et al. 1998). The phenotypic expression of the DNA deletions was development of dark spots on the gray fur of the offspring. Spotting frequency did not increase with an increase in smoke concentration. The investigators reported that chemicals in the particulate phase of cigarette smoke that are possibly responsible for the DNA deletions are B[a]P, cadmium, acetamide, aniline, o-toluidine, acrylonitrile, and catechol. (For a description of transplacental genotoxic effects in rodents, see the section on "Genotoxicity" earlier in this chapter.)
Developmental effects from exposure to cigarette smoke were further studied in the curly-tail mouse and in the C57BL strain, a strain not predisposed to neural tube defects (Seller and Bnait 1995). Mice received nose-only exposure to smoke from commercial low-tar or high-tar cigarettes from day 0 through day 17. Six cigarettes were smoked during each exposure, using one set of smoking-machine conditions (two-second, 35-mL puff). Mice in both treatment groups were sacrificed on day 18, and the embryos were removed and examined for gross congenital malformations. Treated mice (low tar and high tar) showed significant reduction in number of ossification centers in seven regions compared with sham-treated controls. Changes were consistently more marked in the animals exposed to low tar, but the differences were not significantly different from those produced by exposure to smoke from the high-tar cigarettes. One rib abnormality occurred in the C57BL mice, but no major congenital malformations were observed. In the curly-tail mice, a modest increase in the frequency of open spina bifida and exencephaly was observed. The researchers proposed that although cigarette smoke is not a potent teratogen in mice, it may have minor effects in mice that are genetically predisposed to an abnormality.
In a study of pregnancy-related adverse health outcomes from exposure to cigarette smoke, fetuses of Sprague-Dawley rats were examined for abnormalities of the skull, extremities, and other parts of the body (Reznik and Marquard 1980). The exposure regimens varied in the number of exposures per day, in the period of exposure during gestation, and in smoke concentrations from research cigarettes with different yields of nicotine, condensate, and CO. All cigarettes were machine smoked with one set of smoking-machine conditions (two-second puff, one puff per minute, 35-mL puff). None of the regimens produced an increase in malformations.
A study of developmental toxicity in the fetuses of male and female Sprague-Dawley rats exposed to cigarette smoke identified an incomplete supraoccipital ossification and unossified sternebrae significantly more often in smoke-exposed animals than in sham controls (Carmines et al. 2003). The number of skeletal variations was dose dependent. Fetal external and visceral variations in treated animals and controls were not significantly different. The exposure regimen consisted of nose-only inhalation for two hours per day, seven days per week, for four weeks before and during mating for males, and for two weeks before mating, during mating, and through gestation day 20 for females. Three concentrations of mainstream smoke were generated from 1R4F reference cigarettes (two-second puff, one puff per minute, 35-mL puff) by diluting the smoke with filtered, conditioned air. No deaths among male rats were associated with exposure to smoke. Occasional diarrhea, salivation, and red material around the eyes and nose were noted among male rats exposed to smoke and the sham controls. One female rat died of causes unrelated to exposure during the study. Females exposed to cigarette smoke also had diarrhea, salivation, and red material around eyes and nose. The decrease in maternal body weight during gestation days 0 through 20, mean maternal body weight at termination, and mean uterine weight in the group exposed to high smoke concentration (600 mg TPM/m3) were statistically significant compared with those in sham-control female rats.
In another study, two-day-old pups born to Sprague-Dawley rats with nose-only daily exposure to mainstream cigarette smoke from day 2 to day 22 of pregnancy had selective reductions in protein kinase C gamma and delta isoforms and neuronal nitric oxide synthase within the dorsocaudal brainstem, a region relevant to respiratory and other autonomic functions (Hasan et al. 2001). One concentration of cigarette smoke exposure (1,000 mL = 10-mL puff × 10 puffs per cigarette × 10 cigarettes per day at hourly intervals) was used in this study.
Other Health Effects
Immune System
Habitual use of cigarettes results in repeated contact with thousands of chemicals. Researchers have shown that antigens in tobacco and cigarette smoke are capable of stimulating an immune response (Becker et al. 1976; Romanski and Broda 1977; Lehrer et al. 1978, 1980; Francus et al. 1988). Experimental data suggest that nicotine itself can affect the immune system, and at least one researcher has identified an allergic reaction to nicotine in a person exposed to cigarette smoke (Lee et al. 1998; McAllister-Sistilli et al. 1998). In addition to nicotine, other immunologically active chemicals are found in cigarette smoke, including the common additive menthol (Rappaport and Hoffman 1941; McGowan 1966; Becker et al. 1976; Johnson et al. 1990; Mudzinski 1993; Li et al. 1997). Research into mechanisms underlying allergic sensitization induced by cigarette smoke suggests that exposure to cigarette smoke suppresses the normal tolerance to common inhaled allergenic matter (Moerloose et al. 2006). Exposure to ovalbumin, an inert antigen, and mainstream smoke from five unfiltered 2R4F reference cigarettes produced a significant increase in ovalbumin-specific IgE and airway inflammation rich in eosinophils and goblet cells in male Balb/c mice. In mice exposed to ovalbumin and cigarette smoke, levels of cytokine IFN-γ and thymus and activation-regulated chemokine were significantly higher, as were the number of dendritic cells, which are specialized for antigen capture, migration, and T-cell stimulation; activated CD4-positive and CD8-positive T lymphocytes; and peribronchial infiltrates with eosinophils. Mice exposed only to cigarette smoke did not have increased serum IgE, increased total numbers of cells in BAL fluid, goblet cell hyperplasia in lung tissue, or increased levels of cytokines and chemokines in BAL fluid supernatant.
A body of evidence suggests that exposure to cigarette smoke produces changes in cellular and humoral immune function in humans and laboratory animals (Johnson et al. 1990). The immune and host defense systems are highly conserved across species; thus, organs and cells of the immune system in humans, mice, and rats are similar (Selgrade et al. 1995). However, the effect of cigarette smoke on the immune system depends on the species, the duration, and the level of exposure. Short-term, low-level exposures generally do not affect the immune system or may be stimulatory, whereas long-term exposures (six months or more) or high levels of exposure were found to be immunosuppressive (Thomas et al. 1974; Holt et al. 1978; Gregson and Prentice 1981; Sopori et al. 1985; Johnson et al. 1990). Smoking-related changes in the peripheral immune system in humans were observed (Stratton et al. 2001). These changes included high white blood cell counts; high counts of cytotoxic or suppressor T cells; low counts of inducer or helper T cells; slight suppression of T-lymphocyte activity; significantly lower activity of natural killer cells; low titers of circulating immunoglobulin, except for elevated titers of IgE; and increased susceptibility to infection. Researchers observed similar effects in animals. More recently, researchers reported decreased immune response and resistance to transplanted tumor cells in mice with prenatal exposure to cigarettes (Ng et al. 2006).
Animals exposed to cigarette smoke for extended periods were more susceptible to challenges with tumor cells and infectious agents than were unexposed animals (Johnson et al. 1990). Scientists studied male C57BL/6J mice with 26 weeks of exposure to the smoke of a king-size, filter-tipped cigarette, with seven to eight minutes of exposure to the smoke of 30 cigarettes per day on five consecutive days per week and subcutaneous inoculation with tumor cells (Chalmer et al. 1975). Tumors in the mice had a significantly higher mean volume, which is a measure of tumor growth rate, than did unexposed controls. This group also had larger and significantly more lung metastases. Animals exposed for only 10 weeks had a significantly lower mean tumor volume than did control mice. In a study of female C57BL/6 mice, toxic effects to the cellular immune system induced by cigarette smoke resulted in decreased viral neutralization, which was reflected in significant decreases in levels of antibody to serum adenovirus and a decrease in activated CD4 T cells in the lung after adenovirus administration (Robbins et al. 2004). The subchronic daily regimen, which consisted of exposure to mainstream smoke from 1R1 or 1R3 reference cigarettes, also significantly reduced the number of dendritic cells in the lung. Exposure inhibited CD4 T-cell expansion and maximal activation and reduced numbers of activated CD4 and CD8 T cells in response to adenovirus administration. Animals exposed to smoke had percentages of lung macrophages, B cells, and CD4 and CD8 cells similar to those of controls without exposure to cigarette smoke. CD8 cytotoxic T lymphocytes are major effector cells involved in immunologically specific tumor destruction in vivo, and CD4 T cells are essential for controlling CD8 T-cell-dependent eradication of tumors (Shiku 2003).
In another study, tumor cells were injected into offspring of female mice exposed to cigarette smoke and air-only controls (Ng et al. 2006). Litter size, but not body weight of offspring, was significantly reduced by prenatal exposure to cigarette smoke. Male offspring injected with tumor cells at 5 or 10 weeks of age and female offspring injected at 5 weeks had a significant increase in tumor incidence compared with that of offspring of mice exposed to air only. Tumors grew significantly faster in the male offspring with prenatal exposure to cigarette smoke. The scientists observed no treatment-related effect on time to tumor formation. Activity of cytotoxic T lymphocytes in male pups exposed to cigarette smoke was significantly reduced, but no effects on natural killer cell activity, cytokine levels, histology of lymphoid organs, or subpopulations of immune cells were observed. Scientists studied adult mice that were susceptible (A/J strain) or resistant (C3H strain) to lung tumors and were exposed to the tobacco carcinogen NNK (Razani-Boroujerdi and Sopori 2007). The findings suggest that differences in immune response to chemical carcinogens predicted differences in tumor response to the carcinogens. In A/J mice, but not in C3H mice, intraperitoneal treatment with NNK suppressed anti–sheep red blood cell antibody plaque- forming cells; T-cell proliferation induced by concanavalin A; and the rise in intracellular calcium induced by anti–CD3/CD28 antibody. NNK also stimulated a significantly higher expression of cyclooxygenase-2 and of α7 nicotinic acetylcholine receptors in the lungs of A/J mice than in the lungs of C3H mice. The NNK treatments administered in this study resulted in lung tumors in all A/J mice but not in C3H mice.
Subchronic (14 weeks) exposure to a 6-percent concentration of the smoke of filtered medium-tar cigarettes (two-second puff, one puff per minute, 35-mL puff) resulted in increased alveolar macrophage activity in Wistar rats (Gregson and Prentice 1981). The macrophage activity and the increase in levels of macrophage acid phosphatase were dose and time dependent. In a study of Sprague-Dawley rats, exposures of 21 or more weeks to the mainstream smoke of 2R1 reference cigarettes led to significant inhibition of antibody production in lymph node cells associated with the lung (Sopori et al. 1989). Longer exposures of 35 to 39 weeks significantly reduced the plaque-forming response of cells in other lymphoid tissues. The plaque-forming response of lymph node cells associated with the lung to a T-cell-independent antigen was markedly reduced compared with the response of cells from control rats. Proliferative responses of lymphoid tissue associated with the lung to T-cell mitogens were unaffected by this exposure, by the relative amounts of T and B cells in lymph node cells associated with the lung or in the spleen, or by macrophage function in the spleen.
In another study of the immunosuppressive effects of exposure to cigarette smoke in female F-344 rats, chronic, daily whole-body exposures of up to 30 months to mainstream smoke from 1R3 reference cigarettes (two-second puff, two puffs per minute, 70-mL puff) reduced proliferation mediated by T-cell antigen and led to constitutive activation of enzymes involved in activation of the T-cell antigen receptor, tyrosine phosphorylase, and phospholipase C-γ1 (Kalra et al. 2000). At eight months, T-cell proliferation in the spleen was significantly reduced in response to anti–CD3 antibody, which directly binds the T-cell antigen receptor and causes T-cell proliferation in the absence of activation of CD28 on T cells. Other treatment-related evidence of altered antigen-mediated T-cell signaling were depleted calcium stores sensitive to inositol-1,4,5-triphosphate and decreased calcium mobilization in spleen cells after ligation of the T-cell antigen receptor.
Endocrine and Other Effects
Changes in blood glucose were noted in several rodent bioassays. Single but not repeated exposure to mainstream cigarette smoke produced a significant increase in blood glucose levels in anesthetized, mechanically ventilated Sprague-Dawley rats. The smoke was inspired through a tracheal cannula (Iida et al. 1998). In another study, subchronic nose-only inhalation exposure to the mainstream smoke of mentholated or non-mentholated cigarettes (two-second puff, one puff per minute, 35-mL puff) resulted in a significant decrease in blood glucose levels in a high-dose group of F-344 rats exposed to smoke from menthol cigarettes or nonmenthol cigarettes compared with unexposed control animals (Gaworski et al. 1997). Similarly, subchronic nose-only exposure to mainstream smoke from 1R4F reference cigarettes (one puff per minute, 35-mL puff) produced a significant decrease in glucose level in a high-dose group of male Sprague-Dawley rats and in the two groups of female rats with highest doses (Terpstra et al. 2003).
Andersson and colleagues (1985) studied acute, nose-only, intermittent exposure to smoke from one, two, or four unfiltered 1R1 reference cigarettes. This exposure resulted in dose-dependent increases in catecholamine utilization in the dopamine and noradrenaline nerve terminal systems in the hypothalamus of Sprague-Dawley male rats. Luteinizing hormone, prolactin, and thyroid-stimulating hormone were significantly lower in a dose-dependent manner in treated rats than in controls. Corticosterone was significantly increased in rats with the highest exposure. Follicle-stimulating hormone, adrenocorticotropic hormone (ACTH), and vasopressin were not affected by exposure to cigarette smoke. Treated animals received nose-only exposure, but controls were exposed only to air.
In a subsequent study, these investigators reported that, in contrast, acute, nose-only continuous exposure of male Sprague-Dawley rats to the smoke of one, two, or four unfiltered 1R1 reference cigarettes produced smaller reductions in catecholamine levels and increases in catecholamine turnover and did not produce an increase in dopamine utilization in the median eminence (Andersson et al. 1987). The researchers proposed that intermittent exposure to cigarette smoke produced stronger euphoric and neuroendocrine-related effects than did continuous exposure to cigarette smoke. As with male rats, diestrus female Sprague-Dawley rats with intermittent 30 minutes of nose-only exposure to the smoke of one, two, or four unfiltered 1R1 reference cigarettes had decreased catecholamine levels and increased catecholamine utilization in hypothalamic and preoptic noradrenaline nerve terminal systems and decreased serum prolactin and luteinizing hormone (Andersson et al. 1985). The effects were dose and time dependent. In contrast to findings in male rats (Andersson et al. 1985), for female rats, exposure to cigarette smoke caused lower dopamine and noradrena-line levels in the median eminence and lower ACTH levels (Andersson et al. 1988). Exposure to cigarette smoke did not inhibit secretion of the thyroid-stimulating hormone in female rats as it did in male rats. Catecholamine levels were measured in male Sprague-Dawley rats for 48 hours, 72 hours, or 7 days after an exposure regimen that consisted of a daily 2-hour exposure to the smoke of two 1R1 unfiltered reference cigarettes for 10 days (Andersson et al. 1989). At 48 hours after exposure, significantly lower levels of serum corticosterone and serum prolactin were noted and were attributed to maintained activation in dopamine utilization. At 72 hours, serum prolactin levels were still significantly lower than those in controls. Brain regions of increased catecholamine utilization in rats exposed to cigarette smoke decreased with time and were absent by seven days after exposure. Levels of ACTH were not changed relative to those in controls exposed only to air.
Jansson and colleagues (1992) found that age of onset of postnatal endocrine changes varied by the duration of exposure to cigarette smoke. Male Sprague-Dawley rats were exposed daily to the smoke of two 1R1 reference cigarettes, beginning on day 1 after birth and continuing for 5, 10, or 20 days. The rats were sacrificed 24 hours after the 10- or 20-day exposure. Rat pups had a significant increase in serum levels of luteinizing hormone compared with levels in control pups exposed only to air. Animals sacrificed seven months after the 20-day exposure had a significant increase in serum prolactin levels. Pups sacrificed 24 hours after a 20-day exposure had a significant increase in catecholamine utilization in the medial palisade zone of the median eminence and a substantial reduction in catecholamine utilization in the parvocellular and magnocellular parts of the paraventricular hypo-thalamic nucleus. Changes in catecholamine utilization were not seen in animals sacrificed seven months after the 20-day exposure to cigarette smoke. Serum corticosterone levels and dopamine and norepinephrine utilization in the hypothalamus were not significantly different for rats exposed to smoke and controls.
Other researchers noted statistically significant increases in the weight of the adrenal gland relative to body weight in Sprague-Dawley rats after subchronic inhalation exposure to the smoke of 1R4F reference cigarettes (one puff per minute, 35-mL puff) (Terpstra et al. 2003). Compared with the sham controls, the weight of the left adrenal gland increased for males in the two groups with the highest doses, whereas females had an increase in the weight of the left and right adrenal glands in the two groups with the highest doses.
An inverse relationship exists between smoking and body weight in humans, and nicotine is believed to be the chemical mediator (Chen et al. 2005). Direct nicotine administration to humans or animals decreases body weight and caloric intake. Scientists designed a study to determine the effect of short-term exposure to cigarette smoke on appetite control in male Balb/c mice. Inhalation exposure to the smoke of three commercial cigarettes a day for four days led to a significant decrease in plasma concentrations of leptin, a hormone that signals satiety (Chen et al. 2005). Animals exposed to smoke had a decrease in mRNA expression of white adipose tissue UCP1 (a mitochondrial uncoupling protein involved in energy metabolism) and an increase in mRNA expression of brown adipsose tissue UCP3. Food intake and body weight were significantly decreased in the animals exposed to smoke compared with those in the sham controls, even though plasma concentrations of corticosterone were unchanged. Concentrations of hypothalamic neuropeptide Y, which stimulates feeding behavior, were not affected by the acute exposure regimen. Only one concentration of smoke was used in this study, and details on smoking- machine conditions were not provided. Other animal studies with longer durations of exposure to cigarette smoke also documented either weight loss or reduced weight gain in treated animals compared with those in unexposed controls (Ayres et al. 2001; Carmines et al. 2003; Witschi et al. 2004).
Summary
This chapter discusses a wide variety of chemicals found in cigarette smoke. These chemicals extend across a broad spectrum of volatility, lipophilicity, and reactivity, and include compounds that are known or suspected to be carcinogenic, toxic, and addictive. Some of these compounds also promote the carcinogenicity, toxicity, or addictiveness of the other constituents of cigarette smoke. Despite uncertainties about which chemical constituents are responsible for specific adverse health outcomes, there is broad scientific agreement about which chemicals in conventional tobacco-burning cigarettes could be harmful to individuals' health. Less is known about, and research is needed on, the potentially harmful chemicals in smoke from new and emerging cigarette technologies. Cigarette characteristics that influence either nicotine delivery to the smoker or smoke constituents that interact with nicotine deserve special consideration, because nicotine maintains the addiction and thereby leads to ongoing exposures of smokers to chemical compounds with known adverse health effects.
The levels of the chemical constituents in cigarette smoke are influenced by many different factors. The levels of the metals and nitrogen-containing compounds in the tobacco are highly influenced by the soil in which it is grown and the fertilizers used to promote growth of the plant. Many of the chemicals of direct concern vary with the different types of tobacco (e.g., bright, burley, or oriental) that are combined to produce a specific tobacco blend. Within a type of tobacco, the position of the leaf on the stalk can also influence the chemical levels in harvested tobacco leaves that will eventually affect the levels in smoke. The inclusion of reconstituted and expanded tobacco in cigarette fillers can also alter the chemistry of cigarette smoke. After the tobacco is harvested, the method of curing and the addition of humectants, sugars, and flavor-related compounds will change the chemical composition of the tobacco that goes into the cigarette. Different tobacco blends, filters, filter paper, additives, and design innovations employed in cigarette manufacturing have a profound influence on the levels of toxicants transferred from tobacco into the mainstream smoke with every single puff.
It is well documented that cigarettes are not smoked with the same puffing profile. The differences in smoking patterns, including the number of puffs, the puff volume, and whether the smoker blocks the ventilation holes greatly influence the delivery of smoke constituents to the smoker. An individual smoker consumes each cigarette differently, depending on the time of day, on individual stress levels, and on the time since the last nicotine use. The smoker will change the number of puffs taken, the depth of the puff, and the degree to which ventilation holes are blocked, depending on the individual circumstances occurring at that time. In addition, the rate of metabolism of the chemicals after they enter the smoker's body, in addition to other enzymatic and genetic effects, can influence how long the chemical species of concern remains in the smoker's system. It is broadly understood that there is not a single machine-smoking method that can be used to project the levels of chemical constituents that are found in the human body.
Validated biomarkers of exposure that correlate with dose (the number of cigarettes smoked per day) or that provide information on metabolic activation and detoxification have been reported in the literature. Additional research is needed to determine levels of reduction of these chemicals in cigarette smoke that would produce measurable decreases in the dose delivered to the smoker. Although some biomarkers (nicotine and its metabolites and the TSNAs) are specific to tobacco exposure, most are not specific to tobacco and are influenced by diet, occupation, or other environmental factors. Also, although biomarkers typically represent only recent exposures, the strongest determinant of risk for many diseases (e.g., lung cancer) caused by tobacco use is the duration of smoking (IARC 2004). Carcinogen adducts as biomarkers of biologically effective doses are the most direct measure of tobacco-induced damage at cancer sites in smokers. Surrogate measures such as DNA oxidative repair lesions in urine and thioether levels respond in a dose-related manner to exposure to cigarette smoke and reflect an ongoing state of oxidative stress in the body of a smoker. Biomarkers of potential harm exist for all major tobacco-related diseases. The predictive value of these biomarkers is lessened by their nonspecific nature.
In vitro assays using mammalian or bacterial cellular systems show that cigarette smoke is mutagenic and cytotoxic. Genetic damage to the cell and altered metabolic activities probably play a role in tobacco-related chronic diseases such as cancer and cardiovascular disease. Notably, oxidative DNA damage and markers of oxidative stress are represented by increased levels of oxidatively modified DNA bases in urine, white blood cells, and lung tissue and by oxidative damage to sperm DNA and seminal fluid; increased oxidation of cell membrane lipids (F2 − isoprostanes) in adult and cord blood; and decreased levels of reduced glutathione in lung cells and heart tissue. In addition, short-term mutagenicity and cytotoxicity assays have led to the identification of several potentially causative chemical agents in cigarette smoke (e.g., aromatic amines and heterocyclic amine protein pyrolysate products in the Salmonella mutagenicity assay and HCN and acrolein in cytotoxicity assays). Future in vitro research on mutagenicity and cytotoxicity will likely involve cigarette smoke produced under smoking-machine conditions that more closely mimic human smoking behavior, rather than one standard set of conditions such as the FTC or ISO methods.
Many smoking-related effects in humans can be reproduced in experimental animals. Some of the most promising animal models are those for emphysema and cardiovascular disease induced by cigarette smoke. In contrast, animals have not proven to be good models for the type of lung tumors induced by cigarette smoke in humans. In the absence of a widely accepted animal model for tobacco carcinogenesis, ample data show that cigarette smoke and its condensate are tumorigenic in several animal species and are mutagenic in a variety of systems. Current animal studies have attempted to demonstrate a dose-response relationship by using either the smoke or the condensate from one cigarette type diluted to produce several concentrations or the smoke or condensate from cigarettes from different yield categories. In either instance, researchers have used one set of smoking- machine conditions to produce the cigarette smoke or condensate. Standardized smoking-machine conditions such as the FTC or ISO methods are useful for comparisons between cigarettes but are less relevant to the exposure of human smokers. Future studies will likely incorporate alternative smoking-machine conditions required by some countries or designed to mimic human smoking patterns.
In general, an absence of human data requires researchers to use the results of experiments with laboratory animals and nonanimal systems to estimate human risk. The sum of several decades of laboratory research lends experimental support to the epidemiologic observations that cancer, respiratory disease, cardiovascular disease, and other adverse health outcomes are causally related to cigarette smoking. Although some topics were not a primary focus of this chapter, of note are the instances when sidestream smoke, frequently used as a surrogate for environmental tobacco smoke or secondhand smoke, proved to be more toxic than mainstream smoke, which is inhaled directly by the smoker—for example, in the neutral red cytotoxicity assay and in the development of aortic plaque in the cockerel. Many chemicals of concern to public health are present in higher concentrations in sidestream smoke, the main contributor to secondhand smoke exposure, than in mainstream smoke: 1,3-butadiene, ammonia, aromatic amines, benzene, CO, isoprene, nicotine, nitrosamines, PAHs, pyridine, and toluene.
Perhaps the greatest utility of toxicity testing of cigarette smoke and condensate comes from the ability to explore mechanisms by which tobacco and the constituents of its smoke cause disease, to identify better biomarkers of potential disease risk for use in both clinical and population-based studies, and possibly to evaluate the relative contribution of cigarette components and design features (e.g., additives, tobacco blends, nontobacco components, and filter ventilations) to the inherent toxicity and addictiveness of the product.
The uncertainties in understanding all of the factors involved in the delivery and uptake of toxic, carcinogenic, and addictive chemicals in cigarette smoke and the mechanisms of toxicity induced by cigarette smoke should not impede efforts to lower the concentrations of these chemicals in cigarette smoke. There are ways to lower the concentrations of toxic constituents in cigarette smoke, although additional research is needed to determine the levels of reduction required for achievement of measurable and biologically relevant decreases in delivery of these constituents to the smoker. Such approaches include controls over tobacco growing and curing; the types of tobacco used in the filler, including the use of reconstituted tobacco; the use of additives such as menthol; and the design of the cigarette.
Conclusions
-
In spite of uncertainties concerning whether particular cigarette smoke constituents are responsible for specific adverse health outcomes, there is broad scientific agreement that several of the major classes of chemicals in the combustion emissions of burned tobacco are toxic and carcinogenic.
-
The design characteristics of cigarettes, including ventilation features, filters, and paper porosity, have a significant influence on the levels of toxic and carcinogenic chemicals in the inhaled smoke.
-
The different types of tobacco lamina (e.g., bright, burley, or oriental) that are combined to produce a specific tobacco blend have a significant influence on the levels of toxic and carcinogenic chemicals in the combustion emissions of burned tobacco.
-
There is no available cigarette machine-smoking method that can be used to accurately predict doses of the chemical constituents of tobacco smoke received when using tobacco products.
-
Tobacco-specific biomarkers (nicotine and its metabolites and the tobacco-specific nitrosamines) have been validated as quantitative measures of exposure to tobacco smoke among smokers of cigarettes of similar design who do not use other tobacco- containing products.
-
Although biomarkers of potential harm exist for most tobacco-related diseases, many are not specific to tobacco and levels are also influenced by diet, occupation, or other lifestyle or environmental factors.
References
-
Abdallah F. Processing techniques: leaf conditioning, casing, expansion and flavoring. Tobacco Reporter. 2003a;130(7):64–70.
-
Abdallah F. Recon's new role: contributions of a new-generation recon sheet to tobacco products. Tobacco Reporter. 2003b;130(6):58–61.
-
Abel EL. Smoking during pregnancy: a review of effects on growth and development of offspring. Human Biology. 1980;52(4):593–625. [PubMed: 7009384]
-
Adams JD, Lee SJ, Hoffmann D. Carcinogenic agents in cigarette smoke and the influence of nitrate on their formation. Carcinogenesis. 1984;5(2):221–3. [PubMed: 6697439]
-
Adams JD, Lee SJ, Vinchkoski N, Castonguay A, Hoffmann D. On the formation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone during smoking. Cancer Letters. 1983;17(3):339–46. [PubMed: 6831390]
-
Adams JD, O'Mara-Adams J, Hoffmann D. Toxic and carcinogenic agents in undiluted mainstream smoke and sidestream smoke of different types of cigarettes. Carcinogenesis. 1987;8(5):729–31. [PubMed: 3581431]
-
Adamu CA, Bell PF, Mulchi C, Chaney R. Residual metal concentrations in soils and leaf accumulations in tobacco a decade following farmland application of municipal sludge. Environmental Pollution. 1989;56(2):113–26. [PubMed: 15092482]
-
Akehurst BC. Tobacco. 2nd ed. New York: Longman; 1981.
-
Alaoui-Jamali MA, Rossignol G, Schuller HM, Castonguay A. Transplacental genotoxicity of a tobacco-specific N-nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, in Syrian golden hamster. Mutation Research. 1989;223(1):65–72. [PubMed: 2654630]
-
Allen RE, Vickroy DG. The characterization of cigarette smoke from Cytrel smoking products and its comparison to smoke from flue-cured tobacco. III: particulate phase analysis. Beiträge zur Tabakforschung International. 1976;8(7):430–7.
-
Andersen RA, Kasperbauer MJ, Burton HR, Hamilton JL, Yoder EE. Changes in chemical composition of homogenized leaf-cured and air cured Burley tobacco stored in controlled environments. Journal of Agricultural and Food Chemistry. 1982;30(4):663–8.
-
Andersen RA, Kemp TR. Accumulation of 4-(N-methyl-N-nitrosamino)-1-(3-pyridy)-1-butanone in alkaloid genotypes of Burley tobacco during postharvest processing: comparisons with N′-nitrosonornicotine and probable nitrosamine precursors. Cancer Research. 1985;45(11 Pt 1):5287–93. [PubMed: 4053005]
-
Anderson PJ, Wilson JD, Hiller FC. Particle size distribution of mainstream tobacco and marijuana smoke: analysis using the electrical aerosol analyzer. American Review of Respiratory Disease. 1989;140(1):202–5. [PubMed: 2751166]
-
Andersson K, Eneroth P, Fuxe K, Härfstrand A. Effects of acute intermittent exposure to cigarette smoke on hypothalamic and preoptic catecholamine nerve terminal systems and on neuroendocrine function in the diestrus rat. Naunyn-Schmiedeberg's Archives of Pharmacology. 1988;337(2):131–9. [PubMed: 2966898]
-
Andersson K, Fuxe K, Eneroth P, Agnati LF, Härfstrand A. Effects of acute continuous exposure of the rat to cigarette smoke on amine levels and utilization in discrete hypothalamic catecholamine nerve terminal systems and on neuroendocrine function. Naunyn-Schmiedeberg's Archives of Pharmacology. 1987;335(5):521–8. [PubMed: 2886921]
-
Andersson K, Fuxe K, Eneroth P, Jansson A, Härfstrand A. Effects of withdrawal from chronic exposure to cigarette smoke on hypothalamic and preoptic catechol-amine nerve terminal systems and on the secretion of pituitary hormones in the male rat. Naunyn-Schmiedeberg's Archives of Pharmacology. 1989;339(4):387–96. [PubMed: 2739753]
-
Andersson K, Fuxe K, Eneroth P, Mascagni F, Agnati LF. Effects of acute intermittent exposure to cigarette smoke on catecholamine levels and turnover in various types of hypothalamic DA and NA nerve terminal systems as well as on the secretion of adenohypophy-seal hormones and corticosterone. Acta Physiologica Scandinavica. 1985;124(2):277–85. [PubMed: 2861716]
-
Andreoli C, Gigante D, Nunziata A. A review of in vitro methods to assess the biological activity of tobacco smoke with the aim of reducing the toxicity of smoke. Toxicology in Vitro. 2003;17(5–6):587–94. [PubMed: 14599449]
-
Aoshiba K, Tamaoki J, Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2001;281(6):L1392–L1401. [PubMed: 11704535]
-
Artho AJ, Monroe RJ, Weybrew JA. Physical characteristics of cured tobacco. I: simplified procedures for measuring specific volume and fragility. Tobacco Science. 1963;7:191–7.
-
Ashley DL, Beeson MD, Johnson DR, McCraw JM, Richter P, Pirkle JL, Pechacek TF, Song S, Watson CH. Tobacco-specific nitrosamines in tobacco from U.S. brandandnon-U.S.brandcigarettes. Nicotine&Tobacco Research. 2003;5(3):323–31. [PubMed: 12791527]
-
Ashley DL, Bonin MA, Hamar B, McGeehin M. Using the blood concentration of 2,5-dimethylfuran as a marker for smoking. International Archives of Occupational and Environmental Health. 1996;68(3):183–7. [PubMed: 8919847]
-
Atawodi SE, Preussmann R, Spiegelhalder B. Tobacco- specific nitrosamines in some Nigerian cigarettes. Cancer Letters. 1995;97(1):1–6. [PubMed: 7585468]
-
Ayres PH, Hayes JR, Higuchi MA, Mosberg AT, Sagartz JW. Subchronic inhalation by rats of mainstream smoke from a cigarette that primarily heats tobacco compared to a cigarette that burns tobacco. Inhalation Toxicology. 2001;13(2):149–86. [PubMed: 11153066]
-
Babich H, Borenfreund E. Structure-activity relationship (SAR) models established in vitro with the neutral red cytotoxicity assay. Toxicology in Vitro. 1987;1(1):3–9. [PubMed: 20702372]
-
Bache CA, Lisk DJ, Doss GJ, Hoffmann D, Adams JD. Cadmium and nickel in mainstream particulates of cigarettes containing tobacco grown on a low-cadmium soil-sludge mixture. Journal of Toxicology and Environmental Health. 1985;16(3–4):547–52. [PubMed: 4087318]
-
Backhurst JD. Relation between the "strength" of a cigarette and the "extractable nicotine" in the smoke. Brown and Williamson Collection. Research and Development Establishment – Southhampton. 1965. Bates No. 570340734/0758. < http://legacy
.library .ucsf.edu/tid/xud51f00>. -
Baggett MS, Morie GP. Quantitative determination of phenol and alkylphenols in cigarette smoke and their removal by various filters. Tobacco Science. 1973;17:30–2.
-
Baker RR. Product formation mechanisms inside a burning cigarette. Progress in Energy and Combustion Science. 1981;7(2):135–53.
-
Baker RR, Lewis LS. Filter ventilation—has there been a "cover up"? Recent Advances in Tobacco Science. 1997;23:152–96.
-
Baker RR, Massey ED, Smith G. An overview of the effects of tobacco ingredients on smoke chemistry and toxicity. Food and Chemical Toxicology. 2004;42(Suppl):S53–S83. [PubMed: 15072838]
-
Balansky RM, Blagoeva PM. Tobacco smoke-induced clastogenicity in mouse fetuses and in newborn mice. Mutation Research. 1989;223(1):1–6. [PubMed: 2716759]
-
Bartalesi B, Cavarra E, Fineschi S, Lucattelli M, Lunghi B, Martorana PA, Lungarella G. Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. European Respiratory Journal. 2005;25(1):15–22. [PubMed: 15640318]
-
Bassi JA, Rosso P, Moessinger AC, Blanc WA, James LS. Fetal growth retardation due to maternal tobacco smoke exposure in the rat. Pediatric Research. 1984;18(2):127–30. [PubMed: 6199726]
-
Battista SP. In: Wynder EL, Hoffmann D, Gori GB, editors. Cilia toxic components of cigarette smoke; Smoking and Health 1. Modifying the Risk to the Smoker; DHEW Publication No (NIH) 76-1221. Proceedings of the 3rd World Conference on Smoking and Health; June 2–5, 1975; New York, Washington: U.S. Department of Health, Education, and Welfare, Public Health Service, National Institutes of Health, National Cancer Institute; 1976a. pp. 517–34.
-
Battista SP. Ciliatoxicity. Toward Less Hazardous Cigarettes. The First Set of Experimental Cigarettes. Report No. 1. Gori GB, editor. Washington: U.S. Department of Health, Education, and Welfare, Public Health Service, National Institutes of Health; 1976b. pp. 96–103. DHEW Publication No (NIH) 76-905.
-
Becker CG, Dubin T, Wiedemann HP. Hypersensitivity to tobacco antigen. Proceedings of the National Academy of Sciences of the United States of America. 1976;73(5):1712–6. [PMC free article: PMC430370] [PubMed: 1064042]
-
Bell PF, Mulchi CL, Chaney RL. Microelement concentrations in Maryland air-cured tobacco. Communications in Soil Science and Plant Analysis. 1992;23(13–14):1617–28.
-
Bender MA, Preston RJ, Leonard RC, Pyatt BE, Gooch PC, Shelby MD. Chromosomal aberration and sister-chromatid exchange frequencies in peripheral blood lymphocytes of a large human population sample. Mutation Research. 1988;204(3):421–33. [PubMed: 3347214]
-
Bennett CH, Richardson DR. Time-dependent changes in cardiovascular regulation caused by chronic tobacco smoke exposure. Journal of Applied Physiology. 1990;68(1):248–52. [PubMed: 2312465]
-
Benowitz NL, Fitzgerald GA, Wilson M, Zhang Q. Nicotine effects on eicosanoid formation and hemostatic function: comparison of transdermal nicotine and cigarette smoking. Journal of the American College of Cardiology. 1993;22(4):1159–67. [PubMed: 7691912]
-
Benowitz NL, Jacob P III, Fong I, Gupta S. Nicotine metabolic profile in man: comparison of cigarette smoking and transdermal nicotine. Journal of Pharmacology and Experimental Therapeutics. 1994;268(1):296–303. [PubMed: 8301571]
-
Bernert JT Jr, McGuffey JE, Morrison MA, Pirkle JL. Comparison of serum and salivary cotinine measurements by a sensitive high-performance liquid chromatography– tandem mass spectrometry method as an indicator of exposure to tobacco smoke among smokers and nonsmokers. Journal of Analytical Toxicology. 2000;24(5):333–9. [PubMed: 10926356]
-
Bernfeld P, Homburger F. Skin painting studies in Syrian hamsters. Progress in Experimental Tumor Research. 1983;26:128–53. [PubMed: 6405451]
-
Berstein LM, Tsyrlina EV, Gamajunova VB, Bychkova NV, Krjukova OG, Dzhumasultanova SV, Kovalenko IG, Kolesnik OS. Study of tobacco smoke influence on content of estrogens and DNA flow cytometry data in uterine tissue of rats of different age. Hormone and Metabolic Research. 1999;31(1):27–30. [PubMed: 10077346]
-
Bhide SV, Nair J, Maru GB, Nair UJ, Rao BV, Chakraborty MK, Brunnemann KD. Tobacco-specific N-nitrosamines [TSNA] in green mature and processed tobacco leaves from India. Beiträge zur Tabakforschung International. 1987;14(1):29–32.
-
Birner G, Neumann H-G. Biomonitoring of aromatic amines. II: hemoglobin binding of some monocyclic aromatic amines. Archives of Toxicology. 1988;62( 2–3):110–5. [PubMed: 3196145]
-
Blackard CZ. Cigarette design: reconstituted tobacco isn't just cheap recycling, it's a tool for lowering tar and optimizing taste. Tobacco Reporter. 1997;124(10):51–3.
-
Bnait KS, Seller MJ. Ultrastructural changes in 9-day old mouse embryos following maternal tobacco smoke inhalation. Experimental and Toxicologic Pathology. 1995;47(6):453–61. [PubMed: 8871084]
-
Bombick DW, Ayres PH, Doolittle DJ. Cytotoxicity assessment of whole smoke and vapor phase of mainstream and sidestream cigarette smoke from three Kentucky reference cigarettes. Toxicology Methods. 1997a;7(3):177–90.
-
Bombick DW, Bombick BR, Ayres PH, Putnam K, Avalos J, Borgerding MF, Doolittle DJ. Evaluation of the genotoxic and cytotoxic potential of mainstream whole smoke and smoke condensate from a cigarette containing a novel carbon filter. Fundamental and Applied Toxicology. 1997b;39(1):11–7. [PubMed: 9325023]
-
Bombick DW, Putnam K, Doolittle DJ. Comparative cytotoxicity studies of smoke condensates from different types of cigarettes and tobaccos. Toxicology in Vitro. 1998;12(3):241–9. [PubMed: 20654406]
-
Bonassi S, Hagmar L, Strömberg U, Montagud AH, Tinnerberg H, Forni A, Heikkilä P, Wanders S, Wilhardt P, Hansteen I-L, et al. Chromosomal aberrations in lymphocytes predict human cancer independently of exposure to carcinogens. Cancer Research. 2000;60(6):1619–25. [PubMed: 10749131]
-
Bonassi S, Neri M, Lando C, Ceppi M, Lin Y-P, Chang WP, Holland N, Kirsch-Volders M, Zeiger E, Fenech M. Effect of smoking habit on the frequency of micronuclei in human lymphocytes: results from the Human MicroNucleus project. Mutation Research. 2003;543(2):155–66. [PubMed: 12644185]
-
Borgerding MF, Bodnar JA, Wingate DE. The 1999 Massachusetts Benchmark Study: Final Report. Brown & Williamson Collection. 2000. Bates No. 569670588/0712. < http://legacy
.library .ucsf.edu/tid/yek21c00. -
Borgerding M, Klus H. Analysis of complex mixtures—cigarette smoke. Experimental and Toxicologic Pathology. 2005;57(Suppl 1):43–73. [PubMed: 16092717]
-
Borland CDR, Chamberlain AT, Higenbottam TW, Barber RW, Thrush BA. A comparison between the rate of reaction of nitric oxide in the gas phase and in whole cigarette smoke. Beiträge zur Tabakforschung International. 1985;13(2):67–73.
-
Branner B, Kutzer C, Zwickenpflug W, Scherer G, Heller W-D, Richter E. Haemoglobin adducts from aromatic amines and tobacco-specific nitrosamines in pregnant smoking and non-smoking women. Biomarkers. 1998;3(1):35–47. [PubMed: 23899255]
-
Bridges RB, Combs JG, Humble JW, Turbek JA, Rehm SR, Haley NJ. Puffing topography as a determinant of smoke exposure. Pharmacology, Biochemistry, and Behavior. 1990;37(1):29–39. [PubMed: 2263664]
-
Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, et al. Cigarette smoking saturates brain α4β2 nicotinic acetylcholine receptors. Archives of General Psychiatry. 2006;63(8):808–16. [PMC free article: PMC2773659] [PubMed: 16894067]
-
Brougham LR, Cheng H, Pittman KA. Sensitive high-performance liquid chromatographic method for the determination of chlorhexidine in human serum and urine. Journal of Chromatography B: Biomedical Sciences and Applications. 1986;383(2):365–73. [PubMed: 3558566]
-
Brown B, Kolesar J, Lindberg K, Meckley D, Mosberg A, Doolittle D. Comparative studies of DNA adduct formation in mice following dermal application of smoke condensates from cigarettes that burn or primarily heat tobacco. Mutation Research. 1998;414(1–3):21–30. [PubMed: 9630488]
-
Browne CL. The Design of Cigarettes. 3rd ed. Charlotte (NC): Hoechst Celanese Corporation; 1990.
-
Brunnemann KD, Fink W, Moser F. Analysis of volatile N-nitrosamines in mainstream and sidestream smoke from cigarettes by GLC-TEA. Oncology. 1980;37(4):217–22. [PubMed: 7443154]
-
Brunnemann KD, Hoffmann D. The pH of tobacco smoke. Food and Cosmetic Toxicology. 1974;12(1):115–24. [PubMed: 4459223]
-
Brunnemann KD, Hoffmann D. Chemical studies on tobacco smoke. XXXIV: gas chromatographic determination of ammonia in cigarette and cigar smoke. Journal of Chromatographic Sciences. 1975;13(4):159–63. [PubMed: 1127065]
-
Brunnemann KD, Hoffmann D. Decreased concentrations of N-nitrosodiethanolamine and N-nitrosomorpholine in commercial tobacco products. Journal of Agricultural and Food Chemistry. 1991;39(1):207–8.
-
Brunnemann KD, Kagan MR, Cox JE, Hoffmann D. Analysis of 1,3-butadiene and other selected gas-phase components in cigarette mainstream and sidestream smoke by gas chromatography–mass selective detection. Carcinogenesis. 1990;11(10):1863–8. [PubMed: 2208599]
-
Brunnemann KD, Masaryk J, Hoffmann D. Role of tobacco stems in the formation of N-nitrosamines in tobacco and cigarette mainstream and sidestream smoke. Journal of Agricultural and Food Chemistry. 1983;31(6):1221–4.
-
Brunnemann KD, Stahnke G, Hoffmann D. Chemical studies on tobacco smoke. LXI: volatile pyridines: quantitative analysis in mainstream and sidestream smoke of cigarettes and cigars. Analytical Letters. 1978;A11(7):545–60.
-
Brunnemann KD, Yu L, Hoffmann D. Assessment of carcinogenic volatile N-nitrosamines in tobacco and in mainstream and sidestream smoke from cigarettes. Cancer Research. 1977a;37(9):3218–22. [PubMed: 884671]
-
Brunnemann KD, Yu L, Hoffmann D. Chemical studies on tobacco smoke. XLIX: gas chromatographic determination of hydrogen cyanide and cyanogen in tobacco smoke. Journal of Analytical Toxicology. 1977b;1:38–42.
-
Bryant MS, Simmons HF, Harrell RE, Hinson JA. 2,6- Dimethylaniline – hemoglobin adducts from lidocaine in humans. Carcinogenesis. 1994;15(10):2287–90. [PubMed: 7955068]
-
Buckley C, Wyble CW, Borhani M, Ennis TL, Kobayashi DK, Curci JA, Shapiro SD, Thompson RW. Accelerated enlargement of experimental abdominal aortic aneurysms in a mouse model of chronic cigarette smoke exposure. Journal of the American College of Surgeons. 2004;199(6):896–903. [PubMed: 15555973]
-
Buratti M, Pellegrino O, Valla C, Fustinoni S, Brambilla G, Colombi A. Gas chromatography–electron-capture detection of urinary methylhippuric acid isomers as biomarkers of environmental exposure to xylene. Journal of Chromatography B: Biomedical Sciences and Applications. 1999;723(1–2):95–104. [PubMed: 10080637]
-
Burdock GA, editor. Boca Raton (FL): CRC Press; Fenaroli's Handbook of Flavor Additives. (3rd ed) 1995;II:199, 251, 470.
-
Burns DM, Dybing E, Gray N, Hecht S, Anderson C, Sanner T, O'Connor R, Djordjevic M, Dresler C, Hainaut P, et al. Mandated lowering of toxicants in cigarette smoke: a description of the World Health Organization TobReg proposal. Tobacco Control. 2008;17(2):132–41. [PMC free article: PMC2569138] [PubMed: 18375736]
-
Burtin P, Buffe D, Von Kleist S. The carcinoembryonic antigens of human tumours. Triangle. 1972;11(4):123–9. [PubMed: 4572326]
-
Canada Gazette. Tobacco Act: regulations amending the tobacco reporting regulations. P.C. 2005–1126 June 7, 2005. Statutory instruments 2005. Ottawa: Canada Gazette. Part II. 2005 Jun 29;139(13) Wednesday.
-
Caraballo RS, Giovino GA, Pechacek TF, Mowery PD, Richter PA, Strauss WJ, Sharp DJ, Eriksen MP, Pirkle JL, Maurer KR. Racial and ethnic differences in serum cotinine levels of cigarette smokers: Third National Health and Nutrition Examination Survey, 1988–1991. JAMA: the Journal of the American Medical Association. 1998;280(2):135–9. [PubMed: 9669785]
-
Carmines EL. Evaluation of the potential effects of ingredients added to cigarettes. Part 1: cigarette design, testing approach, and review of results. Food and Chemical Toxicology. 2002;40(1):77–91. [PubMed: 11731038]
-
Carmines EL, Gaworski CL, Faqi AS, Rajendran N. In utero exposure to 1R4F reference cigarette smoke: evaluation of developmental toxicity. Toxicological Sciences. 2003;75(1):134–47. [PubMed: 12805648]
-
Carpenter CM, Wayne GF, Pauly JL, Koh HK, Connolly GN. New cigarette brands with flavors that appeal to youth: tobacco marketing strategies. Health Affairs. 2005;24(6):1601–10. [PubMed: 16284034]
-
Carrell RW. α1-Antitrypsin, emphysema and smoking. New Zealand Medical Journal. 1984;97(756):327–8. [PubMed: 6610142]
-
Castell JV, Donato MT, Gómez-Lechón MJ. Metabolism and bioactivation of toxicants in the lung: the in vitro cellular approach. Experimental and Toxicologic Pathology. 2005;57(Suppl 1):189–204. [PubMed: 16092727]
-
Cavarra E, Bartalesi B, Lucattelli M, Fineschi S, Lunghi B, Gambelli F, Ortiz LA, Martorana PA, Lungarella G. Effects of cigarette smoke in mice with different levels of α1-proteinase inhibitor and sensitivity to oxidants. American Journal of Respiratory and Critical Care Medicine. 2001;164(5):886–90. [PubMed: 11549550]
-
Cavusoglu Y, Timuralp B, Us T, Akgün Y, Kudaiberdieva G, Gorenek B, Unalir A, Goktekin O, Ata N. Cigarette smoking increases plasma concentrations of vascular cell adhesion molecule-1 in patients with coronary artery disease. Angiology. 2004;55(4):397–402. [PubMed: 15258685]
-
Centers for Disease Control and Prevention. Prilocaine- induced methemoglobinemia—Wisconsin, 1993. Morbidity and Mortality Weekly Report. 1994;272(18):1403–4. [PubMed: 7933411]
-
Centers for Disease Control and Prevention. Filter ventilation levels in selected U.S. cigarettes, 1997. Morbidity and Mortality Weekly Report. 1997;46(44):1043–7. [PubMed: 9370225]
-
Chalmer J, Holt PG, Keast D. Cell-mediated immune responses to transplanted tumors in mice chronically exposed to cigarette smoke. Journal of the National Cancer Institute. 1975;55(5):1129–34. [PubMed: 1206738]
-
Chamberlain WJ, Baker JL, Chortyk OT, Stephenson MG. Studies on the reduction of nitrosamines in tobacco. Tobacco Science. 1986;30:81–2.
-
Chamberlain WJ, Severson RF, Stephenson MG. Levels of N-nitrosonornicotine in tobaccos grown under varying agronomic conditions. Tobacco Science. 1984;28:156–8.
-
Chang MJ, McDaniel RL, Naworal JD, Self DA. A rapid method for the determination of mercury in mainstream cigarette smoke by two-stage amalgamation cold vapor atomic absorption spectrometry. Journal of Analytical Atomic Spectrometry. 2002;17(7):710–5.
-
Chang MJ, Naworal JD, Walker K, Connell CT. Investigations on the direct introduction of cigarette smoke for trace elements analysis by inductively coupled plasma mass spectrometry. Spectrochimica Acta Part B, Atomic Spectroscopy. 2003;58(11):1979–96.
-
Chen ATL, Reidy JA, Annest JL, Welty TK, Zhou H-G. Increased chromosome fragility as a consequence of blood folate levels, smoking status, and coffee consumption. Environmental and Molecular Mutagenesis. 1989;13(4):319–24. [PubMed: 2737183]
-
Chen H, Vlahos R, Bozinovski S, Jones J, Anderson GP, Morris MJ. Effect of short-term cigarette smoke exposure on body weight, appetite and brain neuropeptide Y in mice. Neuropsychopharmacology. 2005;30(4):713–9. [PubMed: 15508020]
-
Cheng S. Heavy metal pollution in China: origin, pattern and control. Environmental Science and Pollution Research International. 2003;10(3):192–8. [PubMed: 12846382]
-
Chiba M, Masironi R. Toxic and trace elements in tobacco and tobacco smoke. Bulletin of the World Health Organization. 1992;70(2):269–75. [PMC free article: PMC2393306] [PubMed: 1600587]
-
Chortyk OT, Schlotzhauer WS. Studies on the pyrogenesis of tobacco smoke constituents (a review). Beiträge zur Tabakforschung. 1973;7(3):165–78.
-
Churg A, Cherukupalli K. Cigarette smoke causes rapid lipid peroxidation of rat tracheal epithelium. International Journal of Experimental Pathology. 1993;74(2):127–32. [PMC free article: PMC2002122] [PubMed: 8499312]
-
Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-α drives 70% of cigarette smoke–induced emphysema in the mouse. American Journal of Respiratory and Critical Care Medicine. 2004;170(5):492–8. [PubMed: 15184206]
-
Churg A, Zay K, Shay S, Xie C, Shapiro SD, Hendricks R, Wright JL. Acute cigarette smoke–induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. American Journal of Respiratory Cell and Molecular Biology. 2002;27(3):368–74. [PubMed: 12204900]
-
Clark MSG, Rand MJ, Vanov S. Comparison of pharmacological activity of nicotine and related alkaloids occurring in cigarette smoke. Archives of International Pharmacodynamics. 1965;156(2):363–79. [PubMed: 5868944]
-
Coggins CRE. A review of chronic inhalation studies with mainstream cigarette smoke in rats and mice. Toxicologic Pathology. 1998;26(3):307–14. [PubMed: 9608635]
-
Coggins CRE. A minireview of chronic animal inhalation studies with mainstream cigarette smoke. Inhalation Toxicology. 2002;14(10):991–1002. [PubMed: 12396407]
-
Coggins CRE, Ayres PH, Mosberg AT, Sagartz JW, Burger GT, Hayes AW. Ninety-day inhalation study in rats, comparing smoke from cigarettes that heat tobacco with those that burn tobacco. Fundamental and Applied Toxicology. 1989;13(3):460–83. [PMC free article: PMC7130492] [PubMed: 2612779]
-
Cogswell ME, Weisberg P, Spong C. Cigarette smoking, alcohol use and adverse pregnancy outcomes: implications for micronutrient supplementation. Journal of Nutrition. 2003;133(5 Suppl 2):1722S–1731S. [PubMed: 12730490]
-
Collins MH, Moessinger AC, Kleinerman J, Bassi J, Rosso P, Collins AM, James LS, Blanc WA. Fetal lung hypoplasia associated with maternal smoking: a morphometric analysis. Pediatric Research. 1985;19(4):408–12. [PubMed: 4000765]
-
Colquhoun DR, Goldman LR, Cole RN, Gucek M, Mansharamani M, Witter FR, Apelberg BJ, Halden RU. Global screening of human cord blood proteomes for biomarkers of toxic exposure and effect. Environmental Health Perspectives. 2009;117(5):832–8. [PMC free article: PMC2685849] [PubMed: 19478969]
-
Committee on Biological Markers of the National Research Council. Biological markers in environmental health research. Environmental Health Perspectives. 1987;74:3–9. [PMC free article: PMC1474499] [PubMed: 3691432]
-
Connor TH, Ramanujam VMS, Ward JB Jr, Legaor MS. The identification and characterization of a urinary mutagen resulting from cigarette smoke. Mutation Research. 1983;113(2):161–72. [PubMed: 6835243]
-
Counts ME, Hsu FS, Laffoon SW, Dwyer RW, Cox RH. Mainstream smoke constituent yields and predicting relationships from a worldwide market sample of cigarette brands: ISO smoking conditions. Regulatory Toxicology and Pharmacology. 2004;39(2):111–34. [PubMed: 15041144]
-
Counts ME, Morton MJ, Laffoon SW, Cox RH, Lipowicz PJ. Smoke composition and predicting relationships for international commercial cigarettes smoked with three machine-smoking conditions. Regulatory Toxicology and Pharmacology. 2005;41(3):185–227. [PubMed: 15748796]
-
Csalári J, Szántai K. Transfer rate of cadmium, lead, zinc and iron from the tobacco-cut of the most popular Hungarian cigarette brands to the combustion products. Acta Alimentaria. 2002;31(3):279–88.
-
Curry J, Karnaoukhova L, Guenette GC, Glickman BW. Influence of sex, smoking and age on human hprt mutation frequencies and spectra. Genetics. 1999;152(3):1065–77. [PMC free article: PMC1460655] [PubMed: 10388825]
-
Curtin GM, Hanausek M, Walaszek Z, Mosberg AT, Slaga TJ. Short-term in vitro and in vivo analyses for assessing the tumor-promoting potentials of cigarette smoke condensates. Toxicological Sciences. 2004a;81(1):14–25. [PubMed: 15159522]
-
Curtin GM, Higuchi MA, Ayres PH, Swauger JE, Mosberg AT. Lung tumorigenicity in A/J and rasH2 transgenic mice following mainstream tobacco smoke inhalation. Toxicological Sciences. 2004b;81(1):26–34. [PubMed: 15159525]
-
Curvall M, Enzell CR, Jansson T, Pettersson B, Thelestam M. Evaluation of the biological activity of cigarette-smoke condensate fractions using six in vitro short-term tests. Journal of Toxicology and Environmental Health. 1984;14(2–3):163–80. [PubMed: 6502732]
-
Curvall M, Jansson T, Pettersson B, Hedin A, Enzell CR. In vitro studies of biological effects of cigarette smoke condensate. I: genotoxic and cytotoxic effects of neutral, semivolatile constituents. Mutation Research. 1985;157(2–3):169–80. [PubMed: 4022041]
-
Dalbey WE, Nettesheim P, Griesemer R, Caton JE, Guerin MR. In: Sanders CL, Cross FT, Dagle GE, Mahaffey JA, editors. Lifetime exposures of rats to cigarette tobacco smoke; Pulmonary Toxicology of Respirable Particles; Proceedings of the Nineteenth Annual Hanford Life Sciences Symposium; October 22–24, 1979; Spring-field (VA): U.S. Department of Energy; 1980. pp. 522–35.
-
Dalhamn T. In vivo and in vitro ciliotoxic effects of tobacco smoke. Archives of Environmental Health. 1970;21(5):633–4. [PubMed: 5475426]
-
Davis DL. Waxes and lipids in leaf and their relationship to smoking quality and aroma. Recent Advances in Tobacco Science, Vol 2 Leaf Composition and Physical Properties in Relation to Smoking Quality and Aroma; The 30th Tobacco Chemists' Research Conference; Oct. 18, 1976; Brown and Williamson Collection. Bates No. 501010394/0535. < http://legacy
.library .ucsf.edu/tid/vvr10f00>. -
Davis RA, Curvall M. Determination of nicotine and its metabolites in biological fluids: in vivo studies. Analytical Determination of Nicotine and Related Compounds and Their Metabolites. Gorrod JW, Jacob P III, editors. New York: Elsevier Science; 1999. pp. 583–643.
-
de la Chica RA, Ribas I, Giraldo J, Egozcue J, Fuster C. Chromosomal instability in amniocytes from fetuses of mothers who smoke. JAMA: The Journal of the American Medical Association. 2005;293(10):1212–22. [PubMed: 15755944]
-
de Paiva SAR, Zornoff LAM, Okoshi MP, Okoshi K, Cicogna AC, Campana AO. Behavior of cardiac variables in animals exposed to cigarette smoke. Arquivos Brasileiros de Cardiologia. 2003;81(3):225–8. [PubMed: 14569367]
-
DeCaprio AP. Biomarkers: coming of age for environmental health and risk assessment. Environmental Science & Technology. 1997;31(7):1837–48.
-
Del Santo P, Moneti G, Salvadori M, Saltutti C, Delle Rose A, Dolara P. Levels of the adducts of 4-aminobiphenyl to hemoglobin in control subjects and bladder carcinoma patients. Cancer Letters. 1991;60(3):245–51. [PubMed: 1756515]
-
DeMarini DM. Genotoxicity of tobacco smoke and tobacco smoke condensate. Mutation Research. 1983;114(1):59–89. [PubMed: 6219288]
-
DeMarini DM. Genotoxicity of tobacco smoke and tobacco smoke condensate: a review. Mutation Research. 2004;567(2–3):447–74. [PubMed: 15572290]
-
DeMarini DM, Gudi R, Szkudlinska A, Rao M, Recio L, Kehl M, Kirby PE, Polzin G, Richter PA. Genotoxicity of 10 cigarette smoke condensates in four test systems: comparisons between assays and condensates. Mutation Research. 2008;650(1):15–29. [PubMed: 18006367]
-
DeMarini DM, Preston RJ. Smoking while pregnant: trans-placental mutagenesis of the fetus by tobacco smoke. JAMA: The Journal of the American Medical Association. 2005;293(10):1264–5. [PubMed: 15755950]
-
DeMarini DM, Shelton ML, Levine JG. Mutation spectrum of cigarette smoke condensate in Salmonella: comparison to mutations in smoking-associated tumors. Carcinogenesis. 1995;16(10):2535–42. [PubMed: 7586163]
-
Devlin RB, Frampton ML, Ghio AJ. In vitro studies: what is their role in toxicology? Experimental and Toxicologic Pathology. 2005;57(Suppl 1):183–8. [PubMed: 16092726]
-
DiCarlantonio G, Talbot P. Inhalation of mainstream and sidestream cigarette smoke retards embryo transport and slows muscle contraction in oviducts of hamsters (Mesocricetus auratus). Biology of Reproduction. 1999;61(3):651–6. [PubMed: 10456841]
-
Ding YS, Ashley DL, Watson CH. Determination of 10 carcinogenic polycyclic aromatic hydrocarbons in mainstream cigarette smoke. Journal of Agricultural and Food Chemistry. 2007;55(15):5966–73. [PubMed: 17602652]
-
Ding YS, Trommel JS, Yan XJ, Ashley D, Watson CH. Determination of 14 polycyclic aromatic hydrocarbons in mainstream smoke from domestic cigarettes. Environmental Science & Technology. 2005;39(2):471–8. [PubMed: 15707046]
-
Ding YS, Yan XJ, Jain RB, Lopp E, Tavakoli A, Polzin GM, Stanfill SB, Ashley DL, Watson CH. Determination of 14 polycyclic hydrocarbons in mainstream smoke from U.S. brand and non-U.S. brand cigarettes. Environmental Science & Technology. 2006;40(4):1133–8. [PubMed: 16572766]
-
Djordjevic MV, Gay SL, Bush LP, Chaplin JF. Tobacco-specific nitrosamine accumulation and distribution in flue-cured tobacco alkaloid isolines. Journal of Agricultural and Food Chemistry. 1989;37(3):752–6.
-
Djordjevic MV, Stellman SD, Zang E. Doses of nicotine and lung carcinogens delivered to cigarette smokers. Journal of the National Cancer Institute. 2000;92(2):106–11. [PubMed: 10639511]
-
Dobrowsky A. The adsorption of tobacco smoke: how far is a cigarette its own filter? Tobacco Science. 1960;4:126–9.
-
Domino EF. Pharmacological significance of nicotine. Analytical Determination of Nicotine and Related Compounds and Their Metabolites. Gorrod JW, Jacob P III, editors. New York: Elsevier; 1999. pp. 1–11.
-
Dong M, Schmeltz I, Jacobs E, Hoffmann D. Aza-arenes in tobacco smoke. Journal of Analytical Toxicology. 1978;2(1):21–5.
-
Donnelly GM, McKean HE, Heird CS, Green J. Bioassay of a cigarette smoke fraction. I: examination of dose-response relations and dilution bioassay assumptions in a ciliostasis system. Journal of Toxicology and Environmental Health. 1981a;7(3–4):405–17. [PubMed: 7288895]
-
Donnelly GM, McKean HE, Heird CS, Green J. Bioassay of a cigarette smoke fraction. II: experimental design and potency estimations. Journal of Toxicology and Environmental Health. 1981b;7(3–4):419–44. [PubMed: 7288896]
-
Dontenwill W, Chevalier H-J, Harke H-P, Klimisch H-J, Lafrenz U, Reckzeh G, Fleischman B, Keller W. Experimental studies on tumorigenic activity of cigarette smoke condensate on mouse skin. IV: comparative studies of condensates from different reconstituted tobacco leaves, the effect of NaNO3 as additive to tobacco or reconstituted tobacco sheets, the effect of volatile constituents of smoke, the effect of initial treatment with DMBA. Zeitschrift fur Krebsforschung und Klinische Onkologie. 1972;78(3):236–64. [PubMed: 4265007]
-
Doolittle DJ, Winegar R, Lee CK, Caldwell WS, Hayes AW, deBethizy JD. The genotoxic potential of nicotine and its major metabolites. Mutation Research. 1995;344( 3–4):95–102. [PubMed: 7491133]
-
Douben PET, editor. PAHs: An Ecotoxicological Perspective. Hoboken (NJ): John Wiley & Sons; 2003.
-
Drath DB, Shorey JM, Huber GL. Functional and metabolic properties of alveolar macrophages in response to the gas phase of tobacco smoke. Infection and Immunity. 1981;34(1):11–5. [PMC free article: PMC350812] [PubMed: 6271676]
-
Dube MF, Green CR. Methods of collection of smoke for analytical purposes. Recent Advances in Tobacco Science: Formation, Analysis, and Composition of Tobacco Smoke. 1982;8:42–102.
-
Durocher FD. The choice of paper components for low tar cigarettes. Recent Advances in Tobacco Science. 1984;10:52–71.
-
Dwoskin LP, Teng L, Buxton ST, Ravard A, Deo N, Crooks PA. Minor alkaloids of tobacco release [3H]dopamine from superfused rat striatal slices. European Journal of Pharmacology. 1995;276(1–2):195–9. [PubMed: 7781690]
-
Eaker DW. Dynamic behavior and filtration of mainstream smoke in tobacco column and filter. Recent Advances in Tobacco Science. 1990;16:103–87.
-
Eaton DL, Klaassen CD. Principles of toxicology. Casarett & Doull's Toxicology. The Basic Science of Poisons. 6th ed. Klaassen CD, editor. New York: McGraw-Hill; 2001. pp. 11–34.
-
Eccles R. Menthol and related cooling compounds. Journal of Pharmacy and Pharmacology. 1994;46(8):618–30. [PubMed: 7529306]
-
Enzell CF. Terpenoid components of leaf and their relationship to smoking quality and aroma. Recent Advances in Tobacco Science. 1976;2:32–60.
-
Evans WH, Thomas NC, Boardman MC, Nash SJ. Relationships of polycyclic aromatic hydrocarbon yields with particulate matter (water and nicotine free) yields in mainstream and sidestream cigarette smoke. Science of the Total Environment. 1993;136(1–2):101–9.
-
Fairchild A, Colgrove J. Out of the ashes: the life, death, and rebirth of the "safer" cigarette in the United States. American Journal of Public Health. 2004;94(2):192–204. [PMC free article: PMC1448228] [PubMed: 14759927]
-
Federal Trade Commission. Federal Trade Commission Cigarette Report for 2006. Washington: Federal Trade Commission; 2009.
-
Ferri ES, Baratta EJ. Polonium-210 in tobacco products and human tissues. Radiological Health Data Report. 1966;7(9):485–8. [PubMed: 5975012]
-
Finch GL, Nikula KJ, Chen BT, Barr EB, Chang I-Y, Hobbs CH. Effect of chronic cigarette smoke exposure on lung clearance of tracer particles inhaled by rats. Fundamental and Applied Toxicology. 1995;24(1):76–85. [PubMed: 7713345]
-
Fischer S, Castonguay A, Kaiserman M, Spiegelhalder B, Preussmann R. Tobacco-specific nitrosamines in Canadian cigarettes. Journal of Cancer Research and Clinical Oncology. 1990a;116(6):563–8. [PubMed: 2254375]
-
Fischer S, Spiegelhalder B, Eisenbarth J, Preussmann R. Investigations on the origin of tobacco-specific nitrosa-mines in mainstream smoke of cigarettes. Carcinogenesis. 1990b;11(5):723–30. [PubMed: 2335004]
-
Fischer S, Spiegelhalder B, Preussmann R. Influence of smoking parameters on the delivery of tobacco- specific nitrosamines in cigarette smoke—a contribution to relative risk evaluation. Carcinogenesis. 1989a;10(6):1059–66. [PubMed: 2720900]
-
Fischer S, Spiegelhalder B, Preussmann R. Preformed tobacco-specific nitrosamines in tobacco—role of nitrate and influence of tobacco type. Carcinogenesis. 1989b;10(8):1511–7. [PubMed: 2752525]
-
Fischer S, Spiegelhalder B, Preussmann R. Tobacco- specific nitrosamines in mainstream smoke of West German cigarettes—tar alone is not a sufficient index for carcinogenic potential of cigarette smoke. Carcinogenesis. 1989c;10(1):169–73. [PubMed: 2910520]
-
Fischer S, Spiegelhalder B, Preussmann R. Tobacco- specific nitrosamines in European and USA cigarettes. Archiv für Geschwulstforschung. 1990c;60(3):169–76. [PubMed: 2369279]
-
Fisher B. Curing the TSNA problem: major efforts are underway to eliminate tobacco-specific nitrosamines in U.S. flue-cured tobacco. Tobacco Reporter. 2000a;127(8):1–6.
-
Fisher B. Unraveling smoke: as cigarette makers face new challenges, the pressure to understand tobacco smoke is greater than ever. Tobacco Reporter. 2000b;127(9):24–29.
-
Fisher P. Cigarette manufacture. World Agricultural Series Tobacco: Production, Chemistry and Technology. Davis DL, Neilson MT, editors. Ames (IA): Blackwell Publishing; 1999. pp. 350–1.
-
Florek E, Marszalek A. An experimental study of the influences of tobacco smoke on fertility and reproduction. Human & Experimental Toxicology. 1999;18(4):272–8. [PubMed: 10333314]
-
Florek E, Marszalek A, Biczysko W, Szymanowski K. The experimental investigations of the toxic influence of tobacco smoke affecting progeny during pregnancy. Human & Experimental Toxicology. 1999;18(4):245–51. [PubMed: 10333310]
-
Fordyce WB, Hughes IW, Ivinson MG. The filtration of cigarette smoke. Tobacco Science. 1961;5:70–5.
-
Foulds WS, Bronte-Stewart JM, Chisholm IA. Serum thiocyanate concentrations in tobacco amblyopia. Nature. 1968;218(141):586. [PubMed: 5655189]
-
Fowles J, Dybing E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tobacco Control. 2003;12(4):424–30. [PMC free article: PMC1747794] [PubMed: 14660781]
-
Foy JWD, Bombick BR, Bombick DW, Doolittle DJ, Mosberg AT, Swauger JE. A comparison of in vitro toxicities of cigarette smoke condensate from Eclipse cigarettes and four commercially available ultra low-"tar" cigarettes. Food and Chemical Toxicology. 2004;42(2):237–43. [PubMed: 14667470]
-
Francus T, Klein RF, Staiano-Coico L, Becker CG, Siskind GW. Effects of tobacco glycoprotein (TGP) on the immune system. II: TGP stimulates the proliferation of human T cells and the differentiation of human B cells into Ig secreting cells. Journal of Immunology. 1988;140(6):1823–9. [PubMed: 3257988]
-
Frank R, Braun HE, Holdrinet M, Stonefield KI, Elliot JM, Zilkey B, Vickery L, Chang HH. Metal contents and insecticide residues in tobacco soils and cured tobacco leaves collected in southern Ontario. Tobacco Science. 1977;21:74–80.
-
Fréry N, Huel G, Leroy M, Moreau T, Savard R, Blot P, Lellouch J. Vitamin B12 among parturients and their newborns and its relationship with birthweight. European Journal of Obstetrics, Gynecology, and Reproductive Biology. 1992;45(3):155–63. [PubMed: 1511760]
-
Gackowski D, Speina E, Zielinska M, Kowalewski J, Rozalski R, Siomek A, Paciorek T, Tudke B, Olinski R. Products of oxidative DNA damage and repair as possible biomarkers of susceptibility to lung cancer. Cancer Research. 2003;63(16):4899–902. [PubMed: 12941813]
-
Gaworski CL, Dozier MM, Gerhart JM, Rajendran N, Brennecke LH, Aranyi C, Heck JD. 13-Week inhalation toxicity study of menthol cigarette smoke. Food and Chemical Toxicology. 1997;35(7):683–92. [PubMed: 9301652]
-
Gealy R, Zhang L, Siegfried JM, Luketich JD, Keohavong P. Comparison of mutations in the p53 and K-ras genes in lung carcinomas from smoking and nonsmoking women. Cancer Epidemiology, Biomarkers & Prevention. 1999;8(4 Pt 1):297–302. [PubMed: 10207632]
-
Godschalk RWL, Van Schooten F-J, Bartsch H. A critical evaluation of DNA adducts as biological markers for human exposure to polycyclic aromatic compounds. Journal of Biochemistry and Molecular Biology. 2003;36(1):1–11. [PubMed: 12542969]
-
Goldstein JA, Faletto MB. Advances in mechanisms of activation and deactivation of environmental chemicals. Environmental Health Perspectives. 1993;100:169–76. [PMC free article: PMC1519589] [PubMed: 8354165]
-
González E, Monge C, Whittembury J. Ionization constants of 5,5′ – dimethyl – 2,4 – oxazolidinedione (DMO) and nicotine at temperatures and NaCl concentrations of biological interest. Acta Cientifica Venezolana. 1980;31(2):128–30. [PubMed: 7468079]
-
Gordon SM, Wallace LA, Brinkman MC, Callahan PJ, Kenny DV. Volatile organic compounds as breath bio-markers for active and passive smoking. Environmental Health Perspectives. 2002;110(7):689–98. [PMC free article: PMC1240915] [PubMed: 12117646]
-
Graham EA, Croninger AB, Wynder EL. Experimental production of carcinoma with cigarette tar. IV: successful experiments with rabbits. Cancer Research. 1957;17(11):1058–66. [PubMed: 13489709]
-
Gray N, Zaridze D, Robertson C, Krivosheeva L, Sigacheva N, Boyle P. the International Cigarette Variation Group. Variation within global cigarette brands in tar, nicotine, and certain nitrosamines: analytic study. Tobacco Control. 2000;9(3):351. [PMC free article: PMC1748359] [PubMed: 11203247]
-
Green JD, Chalmers J, Kinnard PJ. The transfer of tobacco additives to cigarette smoke: examination of the possible contribution of pyrolysis products to mainstream smoke composition. Beiträge zur Tabakforschung International. 1989;14(5):283–8.
-
Gregson RL, Prentice DE. Aspects of the immunotoxicity of chronic tobacco smoke exposure of the rat. Toxicology. 1981;22(1):23–31. [PubMed: 7336435]
-
Griest WH, Guerin MR. Influence of tobacco type on smoke composition. Recent Advances in Tobacco Science. 1977;3:121–44.
-
Grimmer G, Naujack K-W, Dettbarn G. Gas chromatographic determination of polycyclic aromatic hydrocarbons, aza-arenes, aromatic amines in the particle and vapor phase of mainstream and sidestream smoke of cigarettes. Toxicology Letters. 1987;35(1):117–24. [PubMed: 3810672]
-
Guerassimov A, Hoshino Y, Takubo Y, Turcotte A, Yamamoto M, Ghezzo H, Triantafillopoulos A, Whittaker K, Hoidal JR, Cosio MG. The development of emphysema in cigarette smoke-exposed mice is strain dependent. American Journal of Respiratory and Critical Care Medicine. 2004;170(9):974–80. [PubMed: 15282203]
-
Guerin MR. Chemical composition of cigarette smoke. Banbury Report No 3: A Safe Cigarette? Gori GB, Bock FG, editors. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory; 1980. pp. 191–204.
-
Guerin MR. Formation and physiochemical nature of sidestream smoke. Environmental Carcinogens: Methods of Analysis and Exposure Measurement. Volume 9–Passive Smoking. 81. O'Neill IK, Brunnemann KD, Dodet B, Hoffmann D, editors. Lyon: International Agency for Research on Cancer; IARC Scientific Publications; 1987. pp. 11–23.
-
Guerin MR, Higgins CE, Jenkins RA. Measuring environmental emissions from tobacco combustion: side-stream cigarette smoke literature review. Atmospheric Environment. 1987;21(2):291–7.
-
Guess HE. pH control via weak acid/conjugate base system (common ion effect) in filter. RJ Reynolds Collection. 1980. Bates No. 504168860/8861. < http://legacy
.library .ucsf.edu/tid/ydu58d00>. -
Gupta RC, Arif JM, Gairola CG. Enhancement of pre-existing DNA adducts in rodents exposed to cigarette smoke. Mutation Research. 1999;424(1–2):195–205. [PubMed: 10064861]
-
Harris EM. Cigarette. U.S. Patent. 439,004. 1890. filed Nov. 29, 1889, and issued Oct. 21, 1890.
-
Hasan SU, Simakajornboon N, MacKinnon Y, Gozal D. Prenatal cigarette smoke exposure selectively alters protein kinase C and nitric oxide synthase expression within the neonatal rat brainstem. Neuroscience Letters. 2001;301(2):135–8. [PubMed: 11248441]
-
Hattis DB. The promise of molecular epidemiology for quantitative risk assessment. Risk Analysis. 1986;6(2):181–93. [PubMed: 3615988]
-
Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–4. [PubMed: 9302297]
-
Hecht SS. Human urinary carcinogen metabolites: bio-markers for investigating tobacco and cancer. Carcinogenesis. 2002;23(6):907–22. [PubMed: 12082012]
-
Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nature Reviews. 2003;3(10):733–44. [PubMed: 14570033]
-
Hecht SS. Carcinogenicity studies of inhaled cigarette smoke in laboratory animals: old and new. Carcinogenesis. 2005;26(9):1488–92. [PubMed: 15930027]
-
Hecht SS, Carmella SG, Le K-A, Murphy SE, Li YS, Le C, Jensen J, Hatsukami DK. Effects of reduced cigarette smoking on levels of 1-hydroxypyrene in urine. Cancer Epidemiology, Biomarkers & Prevention. 2004;13(5):834–42. [PubMed: 15159317]
-
Hecht SS, Chen CB, Hirota N, Ornaf RM, Tso TC, Hoffmann D. Tobacco-specific nitrosamines: formation from nicotine in vitro and during tobacco curing and carcinogenicity in strain A mice. Journal of the National Cancer Institute. 1978;60(4):819–24. [PubMed: 633391]
-
Hecht SS, Hoffmann D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis. 1988;9(6):875–84. [PubMed: 3286030]
-
Hecht SS, Murphy SE, Carmella SG, Li S, Jensen J, Le C, Joseph AM, Hatsukami DK. Similar uptake of lung carcinogens by smokers of regular, light, and ultralight cigarettes. Cancer Epidemiology, Biomarkers & Prevention. 2005;14(3):693–8. [PubMed: 15767351]
-
Hecht SS, Ornaf RM, Hoffmann D. Chemical studies on tobacco smoke. XXXIII: N′-nitrosonornicotine in tobacco: analysis of possible contributing factors and biologic implications. Journal of the National Cancer Institute. 1975;54(5):1237–44. [PubMed: 236398]
-
Henningfield JE, Pankow JF, Garrett BE. Ammonia and other chemical base tobacco additives and cigarette nicotine delivery: issues and research needs. Nicotine & Tobacco Research. 2004;6(2):199–205. [PubMed: 15203793]
-
Hind JD. Method of making a reconstituted tobacco sheet. U.S. Patent. 3,386,449. 1968. filed June 16, 1966, and issued June 4, 1968.
-
Hind JD, Seligman RB. Tobacco sheet material. U.S. Patent. 3,353,541. 1967. filed June 16, 1966, and issued Nov. 21, 1967.
-
Hind JD, Seligman RB. Method of preparing a reconstituted tobacco sheet employing a pectin adhesive. U.S. Patent. 3,420,241. 1969. filed June 16, 1966, and issued Jan. 7, 1969.
-
Höfer I, Nil R, Bättig K. Ultralow-yield cigarettes and type of ventilation: the role of ventilation blocking. Pharmacology, Biochemistry, and Behavior. 1991;40(4):907–14. [PubMed: 1816577]
-
Höfer T, Gerner I, Gundert-Remy U, Liebsch M, Schulte A, Spielmann H, Vogel R, Wettig K. Animal testing and alternative approaches for the human health risk assessment under the proposed new European chemicals regulation. Archives of Toxicology. 2004;78(10):549–64. [PubMed: 15170526]
-
Hoffmann D, Adams JD, Brunnemann KD, Hecht SS. Assessment of tobacco-specific N-nitrosamines in tobacco products. Cancer Research. 1979a;39(7 Pt 1):2505–9. [PubMed: 445452]
-
Hoffmann D, Adams JD, Brunnemann KD, Hecht SS. Formation, occurrence, and carcinogenicity of N-nitrosamines in tobacco products. American Chemical Society Symposium Series. 1981;174:247–73.
-
Hoffmann D, Adams JD, Haley NJ. Reported cigarette smoke values: a closer look. American Journal of Public Health. 1983;73(9):1050–3. [PMC free article: PMC1651064] [PubMed: 6881401]
-
Hoffmann D, Adams JD, Wynder EL. Formation and analysis of carbon monoxide in cigarette mainstream and sidestream smoke. Preventive Medicine. 1979b;8(3):344–50. [PubMed: 471953]
-
Hoffmann D, Brunnemann KD. Endogenous formation of N-nitrosoproline in cigarette smokers. Cancer Research. 1983;43(11):5570–4. [PubMed: 6616484]
-
Hoffmann D, Djordjevic MV, Hoffmann I. The changing cigarette. Preventive Medicine. 1997;26(4):427–34. [PubMed: 9245661]
-
Hoffmann D, Dong M, Hecht SS. Origin in tobacco smoke of N′-nitrosonornicotine, a tobacco-specific carcinogen: brief communication. Journal of the National Cancer Institute. 1977;58(6):1841–4. [PubMed: 864762]
-
Hoffmann D, Hecht SS, Ornaf RM, Wynder EL. N′-nitro-sonornicotine in tobacco. Science. 1974;186(4160):265–7. [PubMed: 4414773]
-
Hoffmann D, Hoffmann I. The changing cigarette, 1950–1995. Journal of Toxicology and Environmental Health. 1997;50(4):307–64. [PubMed: 9120872]
-
Hoffmann D, Hoffmann I. Tobacco smoke components [letter] Beiträge zur Tabakforschung International. 1998;18(1):49–52.
-
Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: a controversial issue. A tribute to Ernst L. Wynder. Chemical Research in Toxicology. 2001;14(7):767–90. [PubMed: 11453723]
-
Hoffmann D, Masuda Y, Wynder EL. α-Naphthylamine and β-naphthylamine in cigarette smoke. Nature. 1969;221(5177):254–6. [PubMed: 5763078]
-
Hoffmann D, Wynder EL. Filtration of phenols from cigarette smoke. Journal of the National Cancer Institute. 1963;30:67–84. [PubMed: 13963937]
-
Hoffmann D, Wynder EL. The reduction of the tumor-igenicity of cigarette smoke condensate by addition of sodium nitrate to tobacco. Cancer Research. 1967;27(1):172–4. [PubMed: 6020358]
-
Hoffmann D, Wynder EL. A study of tobacco carcinogenesis. XI: tumor initiators, tumor accelerators, and tumor promoting activity of condensate fractions. Cancer. 1971;27(4):848–64. [PubMed: 5574075]
-
Hoidal JR, Niewoehner DE. Lung phagocyte recruitment and metabolic alterations induced by cigarette smoke in humans and hamsters. American Review of Respiratory Disease. 1982;126(3):548–52. [PubMed: 6289710]
-
Holt PG, Keast D, Mackenzie JS. Immunosuppression in the mouse induced by long-term exposure to cigarette smoke. American Journal of Pathology. 1978;90(1):281–4. [PMC free article: PMC2018231] [PubMed: 619694]
-
Hopkin JM, Tomlinson VS, Jenkins RM. Variation in response to cytotoxicity of cigarette smoke. BMJ (British Medical Journal). 1981;283(6301):1209–11. [PMC free article: PMC1507431] [PubMed: 6797512]
-
Hopkins R, Wood LE, Sinclair NM. Evaluation of methods to estimate cigarette smoke uptake. Clinical Pharmacology and Therapeutics. 1984;36(6):788–95. [PubMed: 6499358]
-
Horak S, Polanska J, Widlak P. Bulky DNA adducts in human sperm: relationship with fertility semen quality, smoking, and environmental factors. Mutation Research. 2003;537(1):53–65. [PubMed: 12742507]
-
Horton AD, Guerin MR. Quantitative determination of sulfur compounds in the gas phase of cigarette smoke. Journal of Chromatography A. 1974;90(1):63–70. [PubMed: 4824663]
-
Hoshino Y, Mio T, Nagai S, Miki H, Ito I, Izumi T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. American Journal of Physiology – Lung Cellular and Molecular Physiology. 2001;281(2):L509–L516. [PubMed: 11435227]
-
Houeto P, Hoffman JR, Got P, Dang Vu B, Baud FJ. Aceto-nitrile as a possible marker of current cigarette smoking. Human & Experimental Toxicology. 1997;16(11):658–61. [PubMed: 9426367]
-
Hsieh Y-S, Chen B-C, Shiow S-J, Wang H-C, Hsu J-D, Wang C-J. Formation of 8-nitroguanine in tobacco cigarette smokers and in tobacco smoke-exposed Wistar rats. Chemico-Biological Interactions. 2002;140(1):67–80. [PubMed: 12044561]
-
Hutt JA, Vuillemenot BR, Barr EB, Grimes MJ, Hahn FF, Hobbs CH, March TH, Gigliotti AP, Seilkop SK, Finch GL, et al. Life-span inhalation exposure to mainstream cigarette smoke induces lung cancer in B6C3F1 mice through genetic and epigenetic pathways. Carcinogenesis. 2005;26(11):1999–2009. [PubMed: 15944214]
-
Iida M, Iida H, Dohi S, Takenaka M, Fujiwara H. Mechanisms underlying cerebrovascular effects of cigarette smoking in rats in vivo. Stroke. 1998;29(8):1656–65. [PubMed: 9707209]
-
Ingebrethsen BJ. Aerosol studies of cigarette smoke. Recent Advances in Tobacco Science: Advances in the Analytical Methodology of Leaf and Smoke. 1986;12:54–142.
-
Ingebrethsen B, Lyman C. Evaporative deposition as a mechanism of sensory stimulation. RJ Reynolds Collection. 1991. Bates No. 508297971/7997. < http://leg-acy
.library .ucsf.edu/tid/jgx93d00>. -
International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Tobacco Smoking. Vol. 38. Lyon (France): International Agency for Research on Cancer; 1986.
-
International Agency for Research on Cancer, IARC Monographs on the Evaluation of the Carcinogenic Risks to Humans: Tobacco Smoke and Involuntary Smoking. Vol. 83. Lyon (France): International Agency for Research on Cancer; 2004. [PMC free article: PMC4781536] [PubMed: 15285078]
-
International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Smokeless Tobacco and Some Tobacco- specific N-Nitrosamines. Vol. 89. Lyon (France): International Agency for Research on Cancer; 2007. [PMC free article: PMC4781254] [PubMed: 18335640]
-
İşcan A, Yiğitoğlu MR, Ece A, Ari Z, Akyildiz M. The effect of cigarette smoking during pregnancy on cord blood lipid, lipoprotein and apolipoprotein levels. Japanese Heart Journal. 1997;38(4):497–502. [PubMed: 9350146]
-
Ishizu Y, Kaneki K, Okada T. A new method to determine the relation between the particle size and chemical composition of tobacco smoke particles. Journal of Aerosol Science. 1987;18(2):123–9.
-
Jacob P III, Yu L, Shulgin AT, Benowitz NL. Minor tobacco alkaloids as biomarkers for tobacco use: comparison of users of cigarettes, smokeless tobacco, cigars, and pipes. American Journal of Public Health. 1999;89(5):731–6. [PMC free article: PMC1508721] [PubMed: 10224986]
-
Jaffe JH, Kanzler M, Friedman L, Stunkard AJ, Verebey K. Carbon monoxide and thiocyanate levels in low tarnicotine smokers. Addictive Behaviors. 1981;6(4):337–44.
-
Jalili T, Murthy GG, Schiestl RH. Cigarette smoke induces DNA deletions in the mouse embryo. Cancer Research. 1998;58(12):2633–8. [PubMed: 9635590]
-
Jansson A, Andersson K, Bjelke B, Eneroth P, Fuxe K. Effects of a postnatal exposure to cigarette smoke on hypothalamic catecholamine nerve terminal systems and on neuroendocrine function in the postnatal and adult male rat: evidence for long-term modulation of anterior pituitary function. Acta Physiologica Scandinavica. 1992;144(4):453–62. [PubMed: 1605047]
-
Jansson T, Curvall M, Hedin A, Enzell CR. In vitro studies of the biological effects of cigarette smoke condensate. III: induction of SCE by some phenolic and related constituents derived from cigarette smoke. A study of structure-activity relationships. Mutation Research. 1988;206(1):17–24. [PubMed: 3412370]
-
Jarmalaite S, Kannio A, Anttila S, Lazutka JR, Husgafvel-Pursiainen K. Aberrant p16 promoter methylation in smokers and former smokers with nonsmall cell lung cancer. International Journal of Cancer. 2003;106(6):913–8. [PubMed: 12918069]
-
Johnson WR. The pyrogenesis and physicochemical nature of tobacco smoke. Recent Advances in Tobacco Science. 1977;3:1–27.
-
Johnson DE, Rhoades JW. N-nitrosamines in smoke condensate from several varieties of tobacco. Journal of the National Cancer Institute. 1972;48(6):1845–7. [PubMed: 5056271]
-
Johnson JD, Houchens DP, Kluwe WM, Craig DK, Fisher GL. Effects of mainstream and environmental tobacco smoke on the immune system in animals and humans: a review. Critical Reviews in Toxicology. 1990;20(5):369–95. [PubMed: 2202327]
-
Johnson WR, Hale RW, Clough SC, Chen PH. Chemistry of the conversion of nitrate nitrogen to smoke products. Nature. 1973a;243(5404):223–5. [PubMed: 4706291]
-
Johnson WR, Hale RW, Nedlock JW, Grubbs HJ, Powell DH. The distribution of products between mainstream and sidestream smoke. Tobacco Science. 1973b;17:141–4.
-
Joseph AM, Hecht SS, Murphy SE, Carmella SG, Le CT, Zhang Y, Han S, Hatsukami DK. Relationships between cigarette consumption and biomarkers of tobacco toxin exposure. Cancer Epidemiology, Biomarkers & Prevention. 2005;14(12):2963–8. [PubMed: 16365017]
-
Kado NY, Manson C, Eisenstadt E, Hsieh DHP. The kinetics of mutagen excretion in the urine of cigarette smokers. Mutation Research. 1985;157(2–3):227–33. [PubMed: 3894961]
-
Kaiserman MJ, Rickert WS. Carcinogens in tobacco smoke: benzo[a]pyrene from Canadian cigarettes and cigarette tobacco. American Journal of Public Health. 1992;82(7):1023–6. [PMC free article: PMC1694072] [PubMed: 1609904]
-
Kalcher K, Kern W, Pietsch R. Cadmium and lead in the smoke of a filter cigarette. Science of the Total Environment. 1993;128(1):21–35. [PubMed: 8424152]
-
Kalra R, Singh SP, Savage SM, Finch GL, Sopori ML. Effects of cigarette smoke on immune response: chronic exposure to cigarette smoke impairs antigen-mediated signaling in T cells and depletes IP3-sensitive Ca2+ stores. Journal of Pharmacology and Experimental Therapeutics. 2000;293(1):166–71. [PubMed: 10734166]
-
Kao-Shan C-S, Fine RL, Whang-Peng J, Lee EC, Chabner BA. Increased fragile sites and sister chromatid exchanges in bone marrow and peripheral blood of young cigarette smokers. Cancer Research. 1987;47(23):6278–82. [PubMed: 3677077]
-
Karube T, Odagiri Y, Takemoto K, Watanabe S. Analyses of transplacentally induced sister chromatid exchanges and micronuclei in mouse fetal liver cells following maternal exposure to cigarette smoke. Cancer Research. 1989;49(13):3550–2. [PubMed: 2731176]
-
Kataoka H, Kijima K, Maruo G. Determination of mutagenic heterocyclic amines in combustion smoke samples. Bulletin of Environmental Contamination and Toxicology. 1998;60(1):60–7. [PubMed: 9484557]
-
Keith CH. Experimental and theoretical aspects of cigarette smoke filtration. American Chemical Society Symposium Series. 1975;17:79–90.
-
Keith CH. Physical mechanisms of smoke filtration. Recent Advances in Tobacco Science. 1978;4:25–45.
-
Keith CH, Derrick JC. Measurement of the particle size distribution and concentration of cigarette smoke by the "conifuge. Tobacco Science. 1960;4:84–91.
-
Keith CH, Norman V, Bates WW Jr. Tobacco smoke filter. U.S. Patent. 3251365. 1966. filed Mar. 4, 1963, and issued May 17, 1966.
-
Keohavong P, Mady HH, Gao WM, Siegfried JM, Luketich JD, Melhem MF. Topographic analysis of K-ras mutations in histologically normal lung tissues and tumours of lung cancer patients. British Journal of Cancer. 2001;85(2):235–41. [PMC free article: PMC2364035] [PubMed: 11461083]
-
Kertsis GD, Sun HH. Philip Morris: increasing the filling power of tobacco lamina filler. Tobacco Reporter. 1984;110(10):92.
-
Khater AEM. Polonium-210 budget in cigarettes. Journal of Environmental Radioactivity. 2004;71(1):33–41. [PubMed: 14557035]
-
Koul A, Singh A, Sandhir R. Effect of α-tocopherol on the cardiac antioxidant defense system and atherogenic lipids in cigarette smoke-inhaling mice. Inhalation Toxicology. 2003;15(5):513–22. [PubMed: 12682861]
-
Kozlowski LT, Mehta NY, Sweeney CT, Schwartz SS, Vogler GP, Jarvis MJ, West RJ. Filter ventilation and nicotine content of tobacco in cigarettes from Canada, the United Kingdom, and the United States. Tobacco Control. 1998;7(4):369–75. [PMC free article: PMC1751464] [PubMed: 10093170]
-
Kozlowski LT, O'Connor RJ, Sweeney CT. Cigarette design, Risks Associated with Smoking Cigarettes with Low Machine-Measured Yields of Tar and Nicotine. Smoking and Tobacco Control Monograph No 13. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 2001. pp. 13–37. NIH Publication No. 02-5047.
-
Kozlowski LT, Rickert WS, Pope MA, Robinson JC, Frekker RC. Estimating the yield to smokers of tar, nicotine, and carbon monoxide from the 'lowest yield' ventilated filter-cigarettes. British Journal of Addiction. 1982;77(2):159–65. [PubMed: 6956362]
-
Kozlowski LT, Sweeny CT, Pillitteri JL. Blocking cigarette filter vent with lips more than doubles carbon monoxide intake from ultra-low tar cigarettes. Experimental and Clinical Psychopharmacology. 1996;4(4):404–8.
-
Kramer AI. R.J. Reynolds: method of expanding tobacco. Tobacco Reporter. 1991;118(9):66.
-
Krause G, Garganta F, Vrieling H, Schere G. Spontaneous and chemically induced point mutations in HPRT cDNA of the metabolically competent human lymphoblas-toid cell line, MCL-5. Mutation Research. 1999;431(2):417–28. [PubMed: 10636005]
-
Krivan V, Schneider G, Baumann H, Reus U. Multi-element characterization of tobacco smoke condensate. Fresenius' Journal of Analytical Chemistry. 1994;348(3):218–25.
-
Kuenemann-Migeot C, Callais F, Momas I, Festy B. Urinary promutagens of smokers: comparison of concentration methods and relation to cigarette consumption. Mutation Research. 1996;368(2):141–7. [PubMed: 8684404]
-
Larson TM, Moring TB, Ireland MS. Nicotine transfer process. U.S. Patent. 4,215,706. 1980. filed Oct. 13, 1978, and issued Aug. 5, 1980.
-
Latha MS, Vijayammal PL, Kurup PA. Effect of exposure of rats to cigarette smoke on the metabolism of lipids. Atherosclerosis. 1988;70(3):225–31. [PubMed: 3365290]
-
LaVoie EJ, Adams JD, Reinhardt J, Rivenson A, Hoffmann D. Toxicity studies on clove cigarette smoke and constituents of clove: determination of LD50 of eugenol by intratracheal installation in rats and hamsters. Archives of Toxicology. 1986;59(2):78–81. [PubMed: 3753194]
-
Lea JS, Coleman R, Kurien A, Schorge JO, Miller DS, Minna JD, Muller CY. Aberrant p16 methylation is a biomarker for tobacco exposure in cervical squamous cell carcinogenesis. American Journal of Obstetrics and Gynecology. 2004;190(3):674–9. [PubMed: 15041998]
-
Lechner JF, Neft RE, Gilliland FD, Crowell RE, Belinsky SA. Molecular identification of individuals at high risk for lung cancer. Radiation Oncology Investigations. 1997;5(3):103–5. [PubMed: 9303064]
-
Lee CK, Brown BG, Reed EA, Hejtmancik M, Mosberg AT, Doolittle DJ, Hayes AW. DNA adduct formation in mice following dermal application of smoke condensates from cigarettes that burn or heat tobacco. Environmental and Molecular Mutagenesis. 1992;20(4):313–9. [PubMed: 1425611]
-
Lee IW, Ahn SK, Choi EH, Lee SH. Urticarial reaction following the inhalation of nicotine in tobacco smoke. British Journal of Dermatology. 1998;138(3):486–8. [PubMed: 9580805]
-
Lefevre PJ. Pharmacology of minor alkaloids of tobacco [French] Journée de la Dépendance Tabagique. 1989;65(40):2424–32.
-
Leffingwell JC. Nitrogen components of leaf and their relationship to smoking quality and aroma. Recent Advances in Tobacco Science. 1976;2:1–31.
-
Lehrer SB, Wilson MR, Salvaggio JE. Immunogenic properties of tobacco smoke. Journal of Allergy and Clinical Immunology. 1978;62(6):368–70. [PubMed: 81845]
-
Lehrer SB, Wilson MR, Salvaggio JE. Immunogenicity of tobacco smoke components in rabbits and mice. International Archives of Allergy and Applied Immunology. 1980;62(1):16–22. [PubMed: 7372361]
-
Leichter J. Growth of fetuses of rats exposed to ethanol and cigarette smoke during gestation. Growth, Development, and Aging. 1989;53(3):129–34. [PubMed: 2599743]
-
Lendvay AT, Laszlo TS. Cigarette peak coal temperature measurements. Beiträge zur Tabakforschung International. 1974;7(5):276–81.
-
Lewis CL. The effect of cigarette construction parameter on smoke generation and yield. Recent Advances in Tobacco Science. 1990;16:73–101.
-
Li Q, Aubrey MT, Christian T, Freed BM. Differential inhibition of DNA synthesis in human T cells by the cigarette tar components hydroquinone and catechol. Fundamental and Applied Toxicology. 1997;38(2):158–65. [PubMed: 9299189]
-
Lichtenbeld H, Vidić B. Effect of maternal exposure to smoke on gas diffusion capacity in neonatal rat. Respiration Physiology. 1989;75(2):129–40. [PubMed: 2711048]
-
Lippman SM, Peters EJ, Wargovich MJ, Stadnyk AN, Dixon DO, Dekmezian RH, Loewy JW, Morice RC, Cunningham JE, Hong KW. Bronchial micronuclei as a marker of an early stage of carcinogenesis in the human tra-cheobronchial epithelium. International Journal of Cancer. 1990;45(5):811–5. [PubMed: 2335384]
-
Löfroth G. Environmental tobacco smoke: overview of chemical composition and genotoxic components. Mutation Research. 1989;222(2):73–80. [PubMed: 2645518]
-
Loft S, Poulsen HE. Estimation of oxidative DNA damage in man from urinary excretion of repair products. Acta Biochimica Polonica. 1998;45(1):133–44. [PubMed: 9701506]
-
Luceri F, Pieraccini G, Moneti G, Dolara P. Primary aromatic amines from side-stream cigarette smoke are common contaminants of indoor air. Toxicology and Industrial Health. 1993;9(3):405–13. [PubMed: 8367883]
-
MacKown CT, Crafts-Brandner SJ, Sutton TG. Tobacco production - relationships among soil nitrate, leaf nitrate, and leaf yield of burley tobacco: effects of nitrogen management. Agronomy Journal. 1999;91(4):613–21.
-
Malaveille C, Vineis P, Estéve J, Ohshima H, Brun G, Hautefeuille A, Gallet P, Ronco G, Terracini B, Bartsch H. Levels of mutagens in the urine of smokers of black and blond tobacco correlate with their risk of bladder cancer. Carcinogenesis. 1989;10(3):577–86. [PubMed: 2924402]
-
Manabe S, Tohyama K, Wada O, Aramaki T. Detection of a carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine (PhIP), in cigarette smoke condensate. Carcinogenesis. 1991;12(10):1945–7. [PubMed: 1934275]
-
Manabe S, Wada O. Carcinogenic tryptophan pyrolysis products in cigarette smoke condensate and cigarette smoke-polluted indoor air. Environmental Pollution. 1990;64(2):121–32. [PubMed: 15092297]
-
Mancini R, Romano G, Sgambato A, Flamini G, Giovagnoli MR, Boninsegna A, Carraro C, Vecchione A, Cittadini A. Polycyclic aromatic hydrocarbon–DNA adducts in cervical smears of smokers and nonsmokers. Gynecologic Oncology. 1999;75(1):68–71. [PubMed: 10502428]
-
Mao L, Lee JS, Kurie JM, Fan YH, Lippman SM, Lee JJ, Ro YJ, Broxson A, Yu R, Morice RC, et al. Clonal genetic alterations in the lungs of current and former smokers. Journal of the National Cancer Institute. 1997;89(12):857–62. [PubMed: 9196251]
-
March TH, Barr EB, Finch GL, Hahn FF, Hobbs CH, Ménache MG, Nikula KJ. Cigarette smoke exposure produces more evidence of emphysema in B6C3F1 mice than in F344 rats. Toxicological Sciences. 1999;51(2):289–99. [PubMed: 10543031]
-
Martonen TB. Deposition patterns of cigarette smoke in human airways. American Industrial Hygiene Association Journal. 1992;53(1):6–18. [PubMed: 1590221]
-
Martonen TB, Musante CJ. Importance of cloud motion on cigarette smoke deposition in lung airways. Inhalation Toxicology. 2000;12(Suppl 4):261–80. [PubMed: 12881896]
-
Matsukura N, Willey J, Miyashita M, Taffe B, Hoffmann D, Waldren C, Puck TT, Harris CC. Detection of direct mutagenicity of cigarette smoke condensate in mammalian cells. Carcinogenesis. 1991;12(4):685–9. [PubMed: 2013132]
-
Mauderly JL, Gigliotti AP, Barr EB, Bechtold WE, Belinksy SA, Hahan FF, Hobbs CA, March TH, Seilkop SK, Finch GL. Chronic inhalation exposure to mainstream cigarette smoke increases lung and nasal tumor incidence in rats. Toxicological Sciences. 2004;81(2):280–92. [PubMed: 15213336]
-
McAllister-Sistilli CG, Caggiula AR, Knopf S, Rose CA, Miller AL, Donny EC. The effects of nicotine on the immune system. Psychoneuroendocrinology. 1998;23(2):175–87. [PubMed: 9621397]
-
McCusker K, Hoidal J. Selective increase of antioxidant enzyme activity in the alveolar macrophages from cigarette smokers and smoke-exposed hamsters. American Review of Respiratory Disease. 1990;141(3):678–82. [PubMed: 2310098]
-
McGowan EM. Menthol urticaria. Archives of Dermatology. 1966;94(1):62–3. [PubMed: 5938225]
-
McRae DD. The physical and chemical nature of tobacco smoke. Recent Advances in Tobacco Science: The Formation and Evolution of Cigarette Smoke. 1990;16:233–323.
-
Meckley D, Hayes JR, Van Kampen KR, Mosberg AT, Swauger JE. A responsive, sensitive, and reproducible dermal tumor promotion assay for the comparative evaluation of cigarette smoke condensates. Regulatory Toxicology and Pharmacology. 2004a;39(2):135–49. [PubMed: 15041145]
-
Meckley DR, Hayes JR, Van Kampen KR, Ayres PH, Mosberg AT, Swauger JE. Comparative study of smoke condensates from 1R4F cigarettes that burn tobacco versus ECLIPSE cigarettes that primarily heat tobacco in the SENCAR mouse dermal tumor promotion assay. Food and Chemical Toxicology. 2004b;42(5):851–63. [PubMed: 15046832]
-
Melikian AA, Djordjevic MV, Chen S, Richie J Jr, Stellman SD. Effect of delivered dosage of cigarette smoke toxins on the levels of urinary biomarkers of exposure. Cancer Epidemiology, Biomarkers & Prevention. 2007a;16(7):1408–15. [PubMed: 17627005]
-
Melikian AA, Djordjevic MV, Hosey J, Zhang J, Chen S, Zang E, Muscat J, Stellman SD. Gender differences relative to smoking behavior and emissions of toxins from mainstream cigarette smoke. Nicotine & Tobacco Research. 2007b;9(3):377–87. [PubMed: 17365769]
-
Melikian AA, Prahalad AK, Hoffmann D. Urinary trans, trans-muconic acid as an indicator of exposure to benzene in cigarette smokers. Cancer Epidemiology, Biomarkers & Prevention. 1993;2(1):47–51. [PubMed: 8420612]
-
Melikian AA, Prahalad AK, Secker-Walker RH. Comparison of the levels of the urinary benzene metabolite trans, trans-muconic acid in smokers and nonsmokers and the effects of pregnancy. Cancer Epidemiology, Biomarkers & Prevention. 1994;3(3):239–44. [PubMed: 8019374]
-
Miller LM, Foster WM, Dambach DM, Doebler D, McKinnon M, Killar L, Longphre M. A murine model of cigarette smoke-induced pulmonary inflammation using intranasally administered smoke-conditioned medium. Experimental Lung Research. 2002;28(6):435–55. [PubMed: 12217211]
-
Milnerowicz H, Zalewski J, Geneja R, Milnerowicz-Nabzdyk E, Zaslawski R, Woyton J. Effects of exposure to tobacco smoke in pregnancies complicated by oligohydraminos and premature rupture of the membranes. I: concentration of Cd and Pb in blood and Zn, Cu, Cd and Pb in amniotic fluid. International Journal of Occupational Medicine and Environmental Health. 2000;13(3):185–93. [PubMed: 11109742]
-
Mitacek EJ, Brunnemann KD, Hoffmann D, Limsila T, Suttajit M, Martin N, Caplan LS. Volatile nitrosamines and tobacco-specific nitrosamines in the smoke of Thai cigarettes: a risk factor for lung cancer and a suspected risk factor for liver cancer in Thailand. Carcinogenesis. 1999;20(1):133–7. [PubMed: 9934860]
-
Moerloose KB, Robays LJ, Maes T, Brusselle GG, Tournoy KG, Joos GF. Cigarette smoke exposure facilitates allergic sensitization in mice. Respiratory Research. 2006;7:49. [PMC free article: PMC1458334] [PubMed: 16571114]
-
Moessinger AC. Mothers who smoke and the lungs of their offspring. Annals of the New York Academy of Sciences. 1989;562:101–4. [PubMed: 2742269]
-
Moore GE, Bock FG. "Tar" and nicotine levels of American cigarettes" Journal of the National Cancer Institute Monographs. 1968;28:89–94. [PubMed: 5671417]
-
Morie GP, Sloan CH. Determination of N-nitrosodimethyl-amine in the smoke of high-nitrate tobacco cigarettes. Beiträge zur Tabakforschung International. 1973;7(2):61–6.
-
Morie GP, Sloan CH, Baggett MS. Parameters affecting the selective filtration of certain tobacco smoke components. Beiträge zur Tabakforschung International. 1975;8(3):145–9.
-
Morrow JD. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(2):279–86. [PubMed: 15591226]
-
Mudzinski SP. Effects of benzo[a]pyrene on concana-valin A-stimulated human peripheral blood mononu-clear cells in vitro: inhibition of proliferation but no effect on parameters related to the G1 phase of the cell cycle. Toxicology and Applied Pharmacology. 1993;119(2):166–74. [PubMed: 8097600]
-
Mulchi CL, Adamu CA, Bell PF, Chaney RL. Residual heavy metal concentrations in sludge-amended coastal plain soils. I: comparison of extractants. Communications in Soil Science and Plant Analysis. 1991;22(9–10):919–41.
-
Mulchi CL, Adamu CA, Bell PF, Chaney RL. Residual heavy metal concentrations in sludge-amended coastal plain soils. II: predicting metal concentrations in tobacco from soil test information. Communications in Soil Science and Plant Analysis. 1992;23(9–10):1053–69.
-
Mulchi CL, Bell PF, Adamu C, Chaney R. Long term availability of metals in sludge amended acid soils. Journal of Plant Nutrition. 1987;10(9–16):1149–61.
-
Muller WJ, Hess GD, Scherer PW. A model of cigarette smoke particle deposition. American Industrial Hygiene Association Journal. 1990;51(5):245–56. [PubMed: 2346112]
-
Muramatsu M. Jackson D. Studies on the Transport Phenomena in Naturally Smoldering Cigarettes. Japan Tobacco Monopoly Research Corporation, Scientific Papers of the Central Research Institute. 1981. Brown and Williamson Collection. Bates No. 650329128/9236. < legacy
.library.ucsf.edu/tid/eyI00f00>. -
Murkovic M. Chemistry, formation and occurrence of genotoxic heterocyclic aromatic amines in fried products. European Journal of Lipid Science and Technology. 2004;106(11):777–85.
-
Murphy SE, Link CA, Jensen J, Le C, Puumala SS, Hecht SS, Carmella SG, Losey L, Hatsukami DK. A comparison of urinary biomarkers of tobacco and carcinogen exposure in smokers. Cancer Epidemiology, Biomarkers & Prevention. 2004;13(10):1617–23. [PubMed: 15466978]
-
Mutation Research. A Nordic data base on somatic chromosome damage in humans. Nordic Study Group on the Health Risk of Chromosome Damage. Mutation Research. 1990;241(3):325–37. [PubMed: 2366811]
-
National Center for Health Statistics. Summary health statistics for US adults: National Health Interview Survey, 2007, Vital and Health Statistics. Series 10, No 240. Hyattsville (MD): U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics; 2007. DHHS Publication No. (PHS) 2008-1563.
-
Navarro J, Causse MB, Desquilbet N, Hervé F, Lallemand D. The vitamin status of low birth weight infants and their mothers. Journal of Pediatric Gastroenterology and Nutrition. 1984;3(5):744–8. [PubMed: 6502374]
-
Newsome JR, Keith CH. Variation of the gas phase composition within a burning cigarette. Tobacco Science. 1965;9:65–9.
-
Ng SP, Silverstone AE, Lai ZW, Zelikoff JT. Effects of pre-natal exposure to cigarette smoke on offspring tumor susceptibility and associated immune mechanisms. Toxicological Sciences. 2006;89(1):135–44. [PubMed: 16207940]
-
Nitsch A, Kalcher K, Greschonig H, Pietsch R. Heavy metals in tobacco smoke. II: trace metals cadmium, lead, copper, cobalt and nickel in Austrian cigarettes and in particle phase and smoke gas. Beiträge zur Tabakforschung International. 1991;15(1):19–32.
-
Norman A. Cigarette design and materials. Tobacco Production, Chemistry, and Technology. Davis DL, Nielsen MT, editors. Malden (MA): Blackwell Science; 1999. pp. 353–87.
-
Norman V. The effect of perforated tipping paper on the yield of various smoke components. Beiträge zur Tabakforschung International. 1974;7(5):282–7.
-
Norman V, Ihrig AM, Larson TM, Moss BL. The effect of some nitrogenous blend components on NO/NOx and HCN levels in mainstream and sidestream smoke. Beiträge zur Tabakforschung International. 1983;12(2):55–62.
-
Norppa H. Genetic susceptibility, biomarker responses, and cancer. Mutation Research. 2003;544(2–3):339–48. [PubMed: 14644336]
-
Obot CJ, Lee KM, Fuciarelli AF, Renne RA, McKinney WJ. Characterization of mainstream cigarette smoke-induced biomarker responses in ICR and C57Bl/6 mice. Inhalation Toxicology. 2004;16(10):701–19. [PubMed: 15371058]
-
Obwegeser R, Oguogho A, Ulm M, Berghammer P, Sinzinger H. Maternal cigarette smoking increases F2-iso-prostanes and reduces prostacyclin and nitric oxide in umbilical vessels. Prostaglandins & Other Lipid Mediators. 1999;57(4):269–79. [PubMed: 10402220]
-
O'Connor RJ, Hurley PJ. Existing technologies to reduce specific toxicant emissions in cigarette smoke. Tobacco Control. 2008;17(Suppl 1):i39–i48. [PubMed: 18768458]
-
Ogushi F, Hubbard RC, Vogelmeier C, Fells GA, Crystal RG. Risk factors for emphysema: cigarette smoking is associated with a reduction in the association rate constant of lung α1-antitrypsin for neutrophil elastase. Journal of Clinical Investigation. 1991;87(3):1060–5. [PMC free article: PMC329901] [PubMed: 1999486]
-
Ohlemiller TJ, Villia KM, Braun E, Eberhardt KR, Harris RH Jr, Lawson JR, Gann RG. Test Methods for Quantifying the Propensity of Cigarettes to Ignite Soft Furnishings. Washington: US Department of Commerce, US National Institute of Standards and Technology; 1993. NIST Special Publication 851.
-
Owen WC, Reynolds ML. The diffusion of gasses through cigarette paper during smoking. Tobacco Science. 1967;11:14–20.
-
Owens WF Jr. Effect of cigarette paper on smoke yield and composition. Recent Advances in Tobacco Science. 1978;4:3–24.
-
Özerol E, Özerol I, Gökdeniz R, Temel I, Akyol O. Effect of smoking on serum concentrations of total homocysteine, folate, vitamin B12, and nitric oxide in pregnancy: a preliminary study. Fetal Diagnosis and Therapy. 2004;19(2):145–8. [PubMed: 14764959]
-
Pakhale SS, Dolas SS, Maru GB. The distribution of total particulate matter (TPM) and nicotine between mainstream and sidestream smoke in bidis and cigarettes. Analytical Letters. 1997;30(2):383–94.
-
Pan X-M, Staprans I, Hardman DA, Rapp JH. Exposure to cigarette smoke delays the plasma clearance of chylo-microns and chylomicron remnants in rats. American Journal of Physiology. 1997;273(1 Pt 1):G158–G163. [PubMed: 9252522]
-
Pan X-M, Staprans I, Read TE, Rapp JH. Cigarette smoke alters chylomicron metabolism in rats. Journal of Vascular Surgery. 1993;18(2):161–9. [PubMed: 8350424]
-
Pankow JF. A consideration of the role of gas/particle partitioning in the deposition of nicotine and other tobacco smoke compounds in the respiratory tract. Chemical Research in Toxicology. 2001;14(11):1465–81. [PubMed: 11712903]
-
Pankow JF, Mader BT, Isabelle LM, Luo W, Pavlick A, Liang C. Conversion of nicotine in tobacco smoke to its volatile and available free-base form through the action of gaseous ammonia. Environmental Science & Technology. 1997;31(8):2428–33.
-
Pankow JF, Tavakoli AD, Luo W, Isabelle LM. Percent free base nicotine in the tobacco smoke particulate matter of selected commercial and reference cigarettes. Chemical Research in Toxicology. 2003;16(8):1014–8. [PubMed: 12924929]
-
Pappas RS, Polzin GM, Watson CH, Ashley DL. Cadmium, lead, and thallium in smoke particulate from counterfeit cigarettes compared to authentic US brands. Food and Chemical Toxicology. 2007;45(2):202–9. [PubMed: 17011104]
-
Pappas RS, Polzin GM, Zhang L, Watson CH, Paschal DC, Ashley DL. Cadmium, lead, and thallium in mainstream tobacco smoke particulate. Food and Chemical Toxicology. 2006;44(5):714–23. [PubMed: 16309811]
-
Park E-M, Park Y-M, Gwak Y-S. Oxidative damage in tissues of rats exposed to cigarette smoke. Free Radical Biology & Medicine. 1998;25(1):79–86. [PubMed: 9655525]
-
Parmeggiani L, editor. Encyclopedia of Occupational Health and Safety. 3rd (revised) ed. Vol. 1. Geneva: International Labour Office; 1983.
-
Parry JM, Parry EM, Johnson G, Quick E, Waters EM. The detection of genotoxic activity and the quantitative and qualitative assessment of the consequences of exposures. Experimental and Toxicologic Pathology. 2005;57(Suppl 1):205–12. [PubMed: 16092728]
-
Parsons LL, Smith MS, Hamilton JL, Mackown CT. Nitrate reduction during curing and processing of Burley tobacco. Tobacco Science. 1986;30:100–3.
-
Patrianakos C, Hoffmann D. Chemical studies on tobacco smoke. LXIV: on the analysis of aromatic amines in cigarette smoke. Journal of Analytical Toxicology. 1979;3(4):150–4.
-
Patskan G, Reininghaus W. Toxicological evaluation of an electrically heated cigarette. Part 1: overview of technical concepts and summary of findings. Journal of Applied Toxicology. 2003;23(5):323–8. [PubMed: 12975771]
-
Pauluhn J. Overview of inhalation exposure techniques: strengths and weaknesses. Experimental and Toxicologic Pathology. 2005;57(Suppl 1):111–28. [PubMed: 16092719]
-
Pavanello S, Simioli P, Carrieri M, Gregorio P, Clonfero E. Tobacco-smoke exposure indicators and urinary mutagenicity. Mutation Research. 2002;521(1–2):1–9. [PubMed: 12437998]
-
Penn RN. Tobacco flavoring: an overview. Perfumer & Fla-vorist. 1997;22:21–8.
-
Penn A, Butler J, Snyder C, Albert RE. Cigarette smoke and carbon monoxide do not have equivalent effects upon development of arteriosclerotic lesions. Artery. 1983;12(2):117–31. [PubMed: 6678580]
-
Penn A, Currie J, Snyder C. Inhalation of carbon monoxide does not accelerate arteriosclerosis in cockerels. European Journal of Pharmacology. 1992;228(2–3):155–64. [PubMed: 1446720]
-
Penn A, Keller K, Snyder C, Nadas A, Chen LC. The tar fraction of cigarette smoke does not promote arteriosclerotic plaque development. Environmental Health Perspectives. 1996;104(10):1108–13. [PMC free article: PMC1469496] [PubMed: 8930554]
-
Penn A, Snyder CA. Arteriosclerotic plaque development is "promoted" by polynuclear aromatic hydrocarbons. Carcinogenesis. 1988;9(12):2185–9. [PubMed: 3142695]
-
Peres AC, Hiromoto G. Evaluation of 210Pb and 210Po in cigarette tobacco produced in Brazil. Journal of Environmental Radioactivity. 2002;62(1):115–9. [PubMed: 12141603]
-
Perfetti TA, Coleman WM III, Smith WS. Determination of mainstream and sidestream cigarette smoke components for cigarettes of different tobacco types and a set of reference cigarettes. Beiträge zur Tabakforschung International. 1998;18(3):95–113.
-
Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21(48):7435–51. [PubMed: 12379884]
-
Pfeifer GP, Hainaut P. On the origin of G→T transversions in lung cancer. Mutation Research. 2003;526(1–2):39–43. [PubMed: 12714181]
-
Phillips DH. DNA adducts as markers of exposure and risk. Mutation Research. 2005;577(1–2):284–92. [PubMed: 15922369]
-
Pillsbury HC, Bright CC, O'Connor KJ, Irish FW. Tar and nicotine in cigarette smoke. Journal of the Association of Official Analytical Chemists. 1969;52(3):458–62.
-
Pinggera G-M, Lirk P, Bodogri F, Herwig R, Steckel-Berger G, Bartsch G, Rieder J. Urinary acetonitrile concentrations correlate with recent smoking behaviour. BJU International. 2005;95(3):306–9. [PubMed: 15679783]
-
Piperi C, Pouli AE, Katerelos NA, Hatzinikolaou DG, Stavridou A, Psallidopoulos MC. Study of the mechanisms of cigarette smoke gas phase cytotoxicity. Anti-cancer Research. 2003;23(3A):2185–90. [PubMed: 12894595]
-
Pittilo RM, Bull HA, Gulati S, Rowles PM, Blow CM, Machin SJ, Woolf N. Nicotine and cigarette smoking: effects on the ultrastructure of aortic endothelium. International Journal of Experimental Pathology. 1990;71(4):573–86. [PMC free article: PMC2002282] [PubMed: 2400739]
-
Pittilo RM, Mackie IJ, Rowles PM, Machin SJ, Woolf N. Effects of cigarette smoking on the ultrastructure of rat thoracic aorta and its ability to produce prostacyclin. Thrombosis and Haemostasis. 1982;48(2):173–6. [PubMed: 6758180]
-
Pluth JM, Ramsey MJ, Tucker JD. Role of maternal exposures and newborn genotypes on newborn chromosome aberration frequencies. Mutation Research. 2000;465(1–2):101–11. [PubMed: 10708975]
-
Polzin GM, Kosa-Maines RE, Ashley DL, Watson CH. Analysis of volatile organic compounds in mainstream cigarette smoke. Environmental Science & Technology. 2007;41(4):1297–1302. [PubMed: 17593733]
-
Polzin GM, Zhang L, Hearn BA, Tavakoli AD, Vaughan C, Ding YS, Ashley DL, Watson CH. Effect of charcoal-containing cigarette filters on gas phase volatile organic compounds in mainstream cigarette smoke. Tobacco Control. 2008;17(Suppl 1):i10–i16. [PubMed: 18768454]
-
Potts RJ, Newbury CJ, Smith G, Notarianni LJ, Jefferies TM. Sperm chromatin damage associated with male smoking. Mutation Research. 1999;423(1–2):103–11. [PubMed: 10029686]
-
Pouli AE, Hatzinikolaou DG, Piperi C, Stavridou A, Psallidopoulos MC, Stavrides JC. The cytotoxic effect of volatile organic compounds of the gas phase of cigarette smoke on lung epithelial cells. Free Radical Biology & Medicine. 2003;34(3):345–55. [PubMed: 12543250]
-
Poulsen HE. Oxidative DNA modifications. Experimental and Toxicologic Pathology. 2005;57(Suppl 1):161–9. [PubMed: 16092724]
-
Prignot J. Quantification and chemical markers of tobacco-exposure. European Journal of Respiratory Disease. 1987;70(1):1–7. [PubMed: 3545884]
-
Prokopczyk B, Hoffmann D, Bologna M, Cunningham AJ, Trushin N, Akerkar S, Boyiri T, Amin S, Desai D, Colosimo S, et al. Identification of tobacco-derived compounds in human pancreatic juice. Chemical Research in Toxicology. 2002;15(5):677–85. [PubMed: 12018989]
-
Putnam KP, Bombick DW, Doolittle DJ. Evaluation of eight in vitro assays for assessing the cytotoxicity of cigarette smoke condensate. Toxicology in Vitro. 2002;16(5):599–607. [PubMed: 12206827]
-
Randerath E, Avitts TA, Reddy MV, Miller RH, Everson RB, Randerath K. Comparative 32P-analysis of cigarette smoke-induced DNA damage in human tissues and mouse skin. Cancer Research. 1986;46(11):5869–77. [PubMed: 3756927]
-
Randerath E, Mittal D, Randerath K. Tissue distribution of covalent DNA damage in mice treated dermally with cigarette 'tar': preference for lung and heart DNA. Carcinogenesis. 1988;9(1):75–80. [PubMed: 2826034]
-
Rappaport BZ, Hoffman MM. Urticaria due to aliphatic aldehydes. JAMA: The Journal of the American Medical Association. 1941;116(24):2656–9.
-
Razani-Boroujerdi S, Sopori ML. Early manifestations of NNK-induced lung cancer: role of lung immunity in tumor susceptibility. American Journal of Respiratory Cell and Molecular Biology. 2007;36(1):13–19. [PMC free article: PMC1899301] [PubMed: 16873770]
-
Reddy MV, Randerath K. A comparison of DNA adduct formation in white blood cells and internal organs of mice exposed to benzo[a]pyrene, dibenzo[c,g]carba-zole, safrole and cigarette smoke condensate. Mutation Research. 1990;241(1):37–48. [PubMed: 2333084]
-
Resnik FE, Houck WG, Geiszler WA, Wickham JE. Factors affecting static burning rate. Tobacco Science. 1977;21:103–7.
-
Reznik G, Marquard G. Effect of cigarette smoke inhalation during pregnancy in Sprague-Dawley rats. Journal of Environmental Pathology and Toxicology. 1980;4(5–6):141–52. [PubMed: 7217842]
-
Rhoades CB Jr, White RT Jr. Mainstream smoke collection by electrostatic precipitation for acid dissolution in a microwave digestion system prior to trace metal determination. Journal of AOAC International. 1997;80(6):1320–31.
-
Rickert W. Today's cigarettes: steps towards reducing the health impact. Nicotine and Public Health. Ferrence R, Slade J, Room R, Pope M, editors. Washington: American Public Health Association; 2000. pp. 135–58.
-
Rickert WS, Collishaw NE, Bray DF, Robinson JC. Estimates of maximum or average cigarette tar, nicotine, and carbon monoxide yields can be obtained from yields under standard conditions. Preventive Medicine. 1986;15(1):82–91. [PubMed: 3714662]
-
Rickert WS, Kaiserman MJ. Levels of lead, cadmium, and mercury in Canadian cigarette tobacco as indicators of environmental change: results of a 21-year study (1968–1988). Environmental Science & Technology. 1994;28(5):924–7. [PubMed: 22191835]
-
Rickert WS, Robinson JC, Bray DF, Rogers B, Collishaw NE. Characterization of tobacco products: a comparative study of the tar, nicotine, and carbon monoxide yields of cigars, manufactured cigarettes, and cigarettes made from fine-cut tobacco. Preventive Medicine. 1985;14(2):226–33. [PubMed: 4048085]
-
Rickert WS, Robinson JC, Collishaw N. Yields of tar, nicotine, and carbon monoxide in the sidestream smoke from 15 brands of Canadian cigarettes. American Journal of Public Health. 1984;74(3):228–31. [PMC free article: PMC1651464] [PubMed: 6696152]
-
Rickert WS, Robinson JC, Collishaw NE. A study of the growth and decay of cigarette smoke NOx in ambient air under controlled conditions. Environment International. 1987;13(6):399–408.
-
Rickert WS, Robinson JC, Collishaw NE, Bray DF. Estimating the hazards of "less hazardous" cigarettes. III: a study of the effect of various smoking conditions on yields of hydrogen cyanide and cigarette tar. Journal of Toxicology and Environmental Health. 1983;12(1):39–54. [PubMed: 6313950]
-
Rickert WS, Trivedi AH, Momin RA, Wright WG, Lauterbach JH. Effect of smoking conditions and methods of collection on the mutagenicity and cytotoxicity of cigarette mainstream smoke. Toxicological Sciences. 2007;96(2):285–93. [PubMed: 17189562]
-
Ritter D, Knebel JW, Aufderheide M. Comparative assessment of toxicities of mainstream smoke from commercial cigarettes. Inhalation Toxicology. 2004;16(10):691–700. [PubMed: 15371057]
-
Riveles K, Roza R, Talbot P. Phenols, quinolines, indoles, benzene, and 2-cyclopenten-1-ones are oviductal toxicants in cigarette smoke. Toxicological Sciences. 2005;86(1):141–51. [PubMed: 15716489]
-
Robbins CS, Dawe DE, Goncharova SI, Pouladi MA, Drannik AG, Swirski FK, Cox G, Stämpfli MR. Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsiveness. American Journal of Respiratory Cell and Molecular Biology. 2004;30(2):202–11. [PubMed: 12920055]
-
Rodgman A, Perfetti TA. The composition of cigarette smoke: a catalogue of the polycyclic aromatic hydrocarbons. Beiträge zur Tabakforschung International. 2006;22(1):13–69.
-
Rodgman A, Perfetti TA. The Chemical Components of Tobacco and Tobacco Smoke. Boca Raton (FL): CRC Press, Taylor & Francis Group; 2009.
-
Rodin SN, Rodin AS. Origins and selection of p53 mutations in lung carcinogenesis. Seminars in Cancer Biology. 2005;15(2):103–12. [PubMed: 15652455]
-
Roemer E, Stabbert R, Rustemeier K, Veltel DJ, Meisgen TJ, Reininghaus W, Carchman RA, Gaworski CL, Podraza KF. Chemical composition, cytotoxicity and mutagenicity of smoke from US commercial and reference cigarettes smoked under two sets of machine smoking conditions. Toxicology. 2004;15;195(1):31–52. [PubMed: 14698566]
-
Romano G, Sgambato A, Boninsegna A, Flamini G, Curigliano G, Yang Q, La Gioia V, Signorelli C, Ferro A, Capelli G, et al. Evaluation of polycyclic aromatic hydrocarbon-DNA adducts in exfoliated oral cells by an immunohistochemical assay. Cancer Epidemiology, Biomarkers & Prevention. 1999;8(1):91–6. [PubMed: 9950245]
-
Romanski B, Broda S. The immunological response to tobacco antigens in the smoker. I: specific precipitins against tobacco antigens in the serum of healthy cigarette smokers. Allergologia et Immunophathologia. 1977;5(6):659–62. [PubMed: 610410]
-
Ronco AM, Arguello G, Munoz L, Gras N, Llanos M. Metals content in placentas from moderate cigarette consumers: correlation with newborn birth weight. Biometals. 2005a;18(3):233–41. [PubMed: 15984568]
-
Ronco AM, Arguello G, Suazo M, Llanos MN. Increased levels of metallothionein in placenta of smokers. Toxicology. 2005b;208(1):133–9. [PubMed: 15664440]
-
Rühl C, Adams JD, Hoffmann D. Chemical studies on tobacco smoke. LXVI: comparative assessment of volatile and tobacco-specific N-nitrosamines in the smoke of selected cigarettes from the U.S.A., West Germany and France. Journal of Analytical Toxicology. 1980;4(5):255–9. [PubMed: 7442138]
-
Sabbioni G, Beyerbach A. Determination of hemoglobin adducts of arylamines in humans. Journal of Chromatography B: Biomedical Sciences and Applications. 1995;667(1):75–83. [PubMed: 7663688]
-
Sakuma H, Kusana M, Munakata S, Ohsumi T, Sugawara S. The distribution of cigarette smoke components between mainstream and sidestream smoke. I: acidic components. Beiträge zur Tabakforschung International. 1983;12(2):63–71.
-
Sakuma H, Kusama M, Yamaguchi K, Sugawara S. The distribution of cigarette smoke components between mainstream and sidestream smoke. III: middle and higher boiling compounds. Beiträge zur Tabakforschung International. 1984;12(5):251–8.
-
Saleh RA, Agarwal A, Sharma RK, Nelson DR, Thomas AJ Jr. Effect of cigarette smoking on levels of seminal oxidative stress in infertile men: a prospective study. Fertility and Sterility. 2002;78(3):491–9. [PubMed: 12215323]
-
Sanchez-Cespedes M, Ahrendt SA, Piantadosi S, Rosell R, Monzo M, Wu L, Westra WH, Yang SC, Jen J, Sidransky D. Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Research. 2001;61(4):1309–13. [PubMed: 11245426]
-
Şardaş S, Walker D, Akyol D, Karakaya AE. Assessment of smoking-induced DNA damage in lymphocytes of smoking mothers of newborn infants using the alkaline single-cell gel electrophoresis technique. Mutation Research. 1995;335(3):213–7. [PubMed: 8524335]
-
Schmeltz I, Hoffmann D. Nitrogen-containing compounds in tobacco and tobacco smoke. Chemical Reviews. 1977;77(3):295–311.
-
Schneider G, Krivan V. Multi-element analysis of tobacco and smoke condensate by instrumental neutron activation analysis and atomic absorption spectrometry. International Journal of Environmental Analytical Chemistry. 1993;53(2):87–100.
-
Scholl TO, Hediger ML, Schall JI, Khoo C-S, Fischer RL. Dietary and serum folate: their influence on the outcome of pregnancy. American Journal of Clinical Nutrition. 1996;63(4):520–5. [PubMed: 8599315]
-
Schur MO, Rickards JC. The design of low yield cigarettes. Tobacco Science. 1960;4:69–77.
-
Schwartz RS, Hecking LT. Determination of geographic origin of agricultural products by multivariate analysis of trace element composition. Journal of Analytical Atomic Spectrometry. 1991;6(8):637–42.
-
Selgrade MJK, Cooper KD, Devlin RB, van Loveren H, Biagini RE, Luster MI. Immunotoxicity—bridging the gap between animal research and human health effects. Fundamental and Applied Toxicology. 1995;24(1):13–21. [PubMed: 7713335]
-
Seller MJ, Bnait KS. Effects of tobacco smoke inhalation on the developing mouse embryo and fetus. Reproductive Toxicology. 1995;9(5):449–59. [PubMed: 8563188]
-
Seller MJ, Bnait KS, Cairns NJ. Effects of maternal tobacco smoke inhalation on early embryonic growth. Effects of Smoking on the Fetus, Neonate, and Child. Poswillo D, Alberman E, editors. New York: Oxford University Press; 1992. pp. 45–59.
-
Shaikh AN, Negi BS, Sadasivan S. Characterization of Indian cigarette tobacco and its smoke aerosol by nuclear and allied techniques. Journal of Radioanalytical and Nuclear Chemistry. 2002;253(2):231–4.
-
Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. American Journal of Pathology. 2003;163(6):2329–35. [PMC free article: PMC1892384] [PubMed: 14633606]
-
Shen H-M, Chia S-E, Ni Z-Y, New A-L, Lee B-L, Ong C-N. Detection of oxidative DNA damage in human sperm and the association with cigarette smoking. Reproductive Toxicology. 1997;11(5):675–80. [PubMed: 9311575]
-
Shephard RJ. Cigarette smoking and reactions to air pollutants. Canadian Medical Association Journal. 1978;118(4):379–81. 383, 392. [PMC free article: PMC1817966] [PubMed: 343900]
-
Shiku H. Importance of CD4+ helper T-cells in antitumor immunity. International Journal of Hematology. 2003;77(5):435–8. [PubMed: 12841380]
-
Sipowicz MA, Amin S, Desai D, Kasprzak KS, Anderson LM. Oxidative DNA damage in tissues of pregnant female mice and fetuses caused by the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Cancer Letters. 1997;117(1):87–91. [PubMed: 9233836]
-
Sithisarankul P, Vineis P, Kang D, Rothman N, Caporaso N, Strickland P. Association of 1-hydroxypyrene-glucuronide in human urine with cigarette smoking and broiled or roasted meat consumption. Biomarkers. 1997;2(4):217–21. [PubMed: 23899213]
-
Skipper PL, Peng X, Soohoo CK, Tannenbaum SR. Protein adducts as biomarkers of human carcinogen exposure. Drug Metabolism Reviews. 1994;26(1–2):111–24. [PubMed: 8082561]
-
Skog KI, Johansson MAE, Jägerstad MI. Carcinogenic heterocyclic amines in model systems and cooked foods: a review on formation, occurrence and intake. Food and Chemical Toxicology. 1998;36(9–10):879–96. [PubMed: 9737435]
-
Skwarzec B, Ulatowski J, Struminska DI, Boryło A. Inhalation of 210Po and 210Pb from cigarette smoking in Poland. Journal of Environmental Radioactivity. 2001;57(3):221–30. [PubMed: 11720371]
-
Smith LE, Denissenko MF, Bennett WP, Li H, Amin S, Tang M-S, Pfeifer GP. Targeting of lung cancer mutational hotspots by polycyclic aromatic hydrocarbons. Journal of the National Cancer Institute. 2000;92(10):803–11. [PubMed: 10814675]
-
Sonnenfeld G, Griffith RB, Hudgens RW. The effect of smoke generation and manipulation variables on the cytotoxicity of mainstream and cigarette sidestream smoke to monolayer cultures of L-929 cells. Archives of Toxicology. 1985;58(2):120–2. [PubMed: 4091656]
-
Sopori ML, Cherian S, Chilukuri R, Shopp GM. Cigarette smoke causes inhibition of the immune response to intratracheally administered antigens. Toxicology and Applied Pharmacology. 1989;97(3):489–99. [PubMed: 2609346]
-
Sopori ML, Gairola CC, DeLucia AJ, Bryant LR, Cherian S. Immune responsiveness of monkeys exposed chronically to cigarette smoke. Clinical Immunology and Immunopathology. 1985;36(3):338–44. [PubMed: 4017293]
-
Spears AW. Selective filtration of volatile phenolic compounds from cigarette smoke. Tobacco Science. 1963;7:76–80.
-
Spiegelhalder B, Bartsch H. Tobacco-specific nitrosamines. European Journal of Cancer Prevention. 1996;5(Suppl 1):33–8. [PubMed: 8972290]
-
Spincer D, Chard BC. The determination of formaldehyde in cigarette smoke. Beiträge zur Tabakforschung International. 1971;6(2):74–8.
-
Stabbert R, Schäfer K-H, Biefel C, Rustemeier K. Analysis of aromatic amines in cigarette smoke. Rapid Communications in Mass Spectrometry. 2003a;17(18):2125–32. [PubMed: 12955743]
-
Stabbert R, Voncken P, Rustemeier K, Haussmann H-J, Roemer E, Schaffernicht H, Patskan G. Toxicological evaluation of an electrically heated cigarette. Part 2: chemical composition of mainstream smoke. Journal of Applied Toxicology. 2003b;23(5):329–39. [PubMed: 12975772]
-
Stanfill SB, Ashley DL. Solid phase microextraction of alkenylbenzenes and other flavor-related compounds from tobacco for analysis by selected ion monitoring gas chromatography–mass spectrometry. Journal of Chromatography A. 1999;858(1):79–89. [PubMed: 10544893]
-
Stanfill SB, Ashley DL. Quantitation of flavor-related alke-nylbenzenes in tobacco smoke particulate by selected ion monitoring gas chromatography-mass spectrometry. Journal of Agricultural and Food Chemistry. 2000;48(4):1298–306. [PubMed: 10775389]
-
Steegers-Theunissen RP, Van Iersel CA, Peer PG, Nelen WL, Steegers EA. Hyperhomocysteinemia, pregnancy complications, and the timing of investigation. Obstetrics and Gynecology. 2004;104(2):336–43. [PubMed: 15292008]
-
Steele RH, Payne VM, Fulp CW, Rees DC, Lee CK, Doolittle DJ. A comparison of the mutagenicity of the mainstream cigarette smoke condensates from a representative sample of the U.S. cigarette market with a Kentucky reference cigarette (K1R4F). Mutation Research. 1995;342(3–4):179–90. [PubMed: 7715619]
-
Stephens WE, Calder A, Newton J. Source and health implications of high toxic metal concentrations in illicit tobacco products. Environmental Science & Technology. 2005;39(2):479–88. [PubMed: 15707047]
-
Stewart CA, Lawrence BM. Project XGT: effects of levulinic acid, tobacco essence and nicotine salts on smoke pH. Project 0113-1988. RJ Reynolds Collection. Bates No.507869978/9986.< http://legacy
.library .ucsf.edu/tid/ggk14d00>. -
Stober W. Generation, size distribution and composition of tobacco smoke aerosols. Recent Advances in Tobacco Science: Formation, Analysis, and Composition of Tobacco Smoke. 1982;8:3–41.
-
Stratton K, Shetty P, Wallace R, Bonderant S, editors. Clearing the Smoke: Assessing the Science Base for Tobacco Harm Reduction. Washington: National Academy Press; 2001. [PubMed: 25057541]
-
Subramaniam S, Bummer P, Gairola CG. Biochemical and biophysical characterization of pulmonary surfactant in rats exposed chronically to cigarette smoke. Fundamental and Applied Toxicology. 1995;27(1):63–9. [PubMed: 7589929]
-
Sugimura T. Overview of carcinogenic heterocyclic amines. Mutation Research. 1997;376(1–2):211–9. [PubMed: 9202758]
-
Sugimura T, Nagao M, Kawachi T, Honda M, Yahagi T, Seino Y, Sato S, Matsukura N, Matsushima T, Shirai A, et al. Mutagen-carcinogens in food, with special reference to highly mutagenic pyrolytic products in broiled foods. Origins of Human Cancer. Book C, Human Risk Assessment, Section 17. Hiatt HH, Watson JD, Winsten JA, editors. New York: Cold Spring Harbor Press; 1977. pp. 1561–77.
-
Suzuki T, Shishido S, Urushiyama K. Mercury in cigarettes. Tohoku Journal of Experimental Medicine. 1976;119(4):353–6. [PubMed: 960100]
-
Sweeney CT, Kozlowski LT, Parsa P. Effect of filter vent blocking on carbon monoxide exposure from selected lower tar cigarette brands. Pharmacology, Biochemistry, and Behavior. 1999;63(1):167–73. [PubMed: 10340538]
-
Takubo Y, Guerassimov A, Ghezzo H, Triantafillopoulos A, Bates JHT, Hoidal JR, Cosio MG. α1-antitrypsin determines the pattern of emphysema and function in tobacco smoke–exposed mice: parallels with human disease. American Journal of Respiratory and Critical Care Medicine. 2002;166(12 Pt 1):1596–603. [PubMed: 12471075]
-
Talaska G, Schamer M, Skipper P, Tannenbaum S, Caporaso N, Unruh L, Kadlubar FF, Bartsch H, Malaveille C, Vineis P. Detection of carcinogen-DNA adducts in exfoliated urothelial cells of cigarette smokers: association with smoking, hemoglobin adducts, and urinary mutagenicity. Cancer Epidemiology, Biomarkers & Prevention. 1991;1(1):61–6. [PubMed: 1845172]
-
Talbot P, DiCarlantonio G, Knoll M, Gomez C. Identification of cigarette smoke components that alter functioning of hamster (Mesocricetus auratus) oviducts in vitro. Biology of Reproduction. 1998;58(4):1047–53. [PubMed: 9546738]
-
Tani S, Dimayuga PC, Anazawa T, Chyu K-Y, Li H, Shah PK, Cercek B. Aberrant antibody responses to oxidized LDL and increased intimal thickening in apoE−/− mice exposed to cigarette smoke. Atherosclerosis. 2004;175(1):7–14. [PubMed: 15186941]
-
Teague CE Jr. Implications and activities arising from correlation of smoke pH with nicotine impact, other smoke qualities, and cigarette sales. RJ Reynolds Collection. 1974. Bates No. 511223463/3484. < http://legacy
.library .ucsf.edu/tid/rte53d00>. -
Telišman S, Jurasović J, Pizent A, Cvitković P. Cadmium in the blood and seminal fluid of nonoccupationally exposed adult male subjects with regard to smoking habits. International Archives of Occupational and Environmental Health. 1997;70(4):243–8. [PubMed: 9342624]
-
Terpstra PM, Teredesai A, Vanscheeuwijck PM, Verbeeck J, Schepers G, Radtke F, Kuhl P, Gomm W, Anskeit E, Patskan G. Toxicological evaluation of an electrically heated cigarette. Part 4: subchronic inhalation toxicology. Journal of Applied Toxicology. 2003;23(5):349–62. [PubMed: 12975774]
-
Tewes FJ, Meisgen TJ, Veltel DJ, Roemer E, Patskan G. Toxicological evaluation of an electrically heated cigarette. Part 3: genotoxicity and cytotoxicity of mainstream smoke. Journal of Applied Toxicology. 2003;23(5):341–8. [PubMed: 12975773]
-
Thayer PS. Inhibition of cell culture. Toward Less Hazardous Cigarettes. The First Set of Experimental Cigarettes. Report No. 1. Gori GB, editor. Washington: U.S. Department of Health, Education, and Welfare, Public Health Service, National Institutes of Health, National Cancer Institute, Smoking and Health Program; 1976a. pp. 107–8. DHEW Publication No (NIH) 76-905.
-
Thayer PS. Inhibition of macrophages. Toward Less Hazardous Cigarettes. The First Set of Experimental Cigarettes. Report No. 1. Gori GB, editor. Washington: U.S. Department of Health, Education, and Welfare, Public Health Service, National Institutes of Health, National Cancer Institute, Smoking and Health Program; 1976b. pp. 104–6. DHEW Publication No (NIH) 76-905.
-
Thayer PS, Kensler CJ. Cigarette smoke: charcoal filters reduce components that inhibit growth of cultured human cells. Science. 1964;146(3644):642–4. [PubMed: 14191700]
-
Theophilus EH, Bombick BR, Meckley DR, Higuchi MA, Borgerding MF, Morton MJ, Mosberg AT, Swauger JE. Toxicological evaluation of propane expanded tobacco. Food and Chemical Toxicology. 2003a;41(12):1771–80. [PubMed: 14563402]
-
Theophilus EH, Pence DH, Meckley DR, Higuchi MA, Bombick BR, Borgerding MF, Ayres PH, Swauger JE. Toxicological evaluation of expanded shredded tobacco stems. Food and Chemical Toxicology. 2004;42(4):631–9. [PubMed: 15019188]
-
Theophilus EH, Poindexter DB, Meckley DR, Bombick BR, Borgerding MF, Higuchi MA, Ayres PH, Morton MJ, Mosberg AT, Swauger JE. Toxicological evaluation of dry ice expanded tobacco. Toxicology Letters. 2003b;145(2):107–19. [PubMed: 14581163]
-
Thier R, Lewalter J, Selinski S, Bolt HM. Re-evaluation of the effect of smoking on the methylation of N-terminal valine in haemoglobin. Archives of Toxicology. 2001;75(5):270–3. [PubMed: 11548119]
-
Thomas WR, Holt PG, Keast D. Antibody production in mice chronically exposed to fresh cigarette smoke. Experientia. 1974;30(12):1469–70. [PubMed: 4613568]
-
Tomkins BA, Jenkins RA, Griest WH, Reagan RR, Holladay SK. Liquid chromatography determination of benzo[a] pyrene in total particulate matter of cigarette smoke. Journal of the Association of Official Analytical Chemists. 1985;68(5):935–40. [PubMed: 4055640]
-
Torikai K, Uwano Y, Nakamori T, Tarora W, Takahashi H. Study on tobacco components involved in the pyrolytic generation of selected smoke constituents. Food Chemistry and Toxicology. 2005;43(4):559–68. [PubMed: 15721203]
-
Torikai K, Yoshida S, Takahashi H. Effects of temperature, atmosphere and pH on the generation of smoke compounds during tobacco pyrolysis. Food and Chemical Toxicology. 2004;42(9):1409–17. [PubMed: 15234071]
-
Torjussen W, Zachariasen H, Andersen I. Cigarette smoking and nickel exposure. Journal of Environmental Monitoring. 2003;5(2):198–201. [PubMed: 12729253]
-
Torrence KM, McDaniel RL, Self DA, Chang MJ. Slurry sampling for the determination of arsenic, cadmium, and lead in mainstream cigarette smoke condensate by graphite furnace–atomic absorption spectrometry and inductively coupled plasma–mass spectrometry. Analytical and Bioanalytical Chemistry. 2002;372( 5–6):723–31. [PubMed: 11941445]
-
Touey GP, Mumpower RC. Measurement of the combustion-zone temperature of cigarettes. Tobacco Science. 1957;1:33–7.
-
Tretyakova N, Matter B, Jones R, Shallop A. Formation of benzo[a]pyrene diol epoxide–DNA adducts at specific guanines within K-ras and p53 gene sequences: stable isotope-labeling mass spectrometry approach. Biochemistry. 2002;41(30):9535–44. [PubMed: 12135376]
-
Tricker AR, Ditrich C, Preussmann R. N-nitroso compounds in cigarette tobacco and their occurrence in mainstream tobacco smoke. Carcinogenesis. 1991;12(2):257–61. [PubMed: 1995191]
-
Tso TC. Ch. 21–Organic metabolism–alkaloids, Production and Biochemistry of Tobacco Plant. Beltsville (MD): Ideals, Inc; 1990.
-
Tso TC, Chaplin JF, Adams JD, Hoffmann D. Simple correlation and multiple regression among leaf and smoke characteristics of burley tobaccos. Beiträge zur Tabakforschung International. 1982;11(3):141–50.
-
Tso TC, Sims JL, Johnson DE. Some agronomic factors affecting N-dimethylnitrosamine content in cigarette smoke. Beiträge zur Tabakforschung International. 1975;8(1):34–8.
-
Tsuji T, Aoshiba K, Nagai A. Cigarette smoke induces senescence in alveolar epithelial cells. American Journal of Respiratory Cell and Molecular Biology. 2004;31(6):643–9. [PubMed: 15333326]
-
Tuomisto J, Kolonen S, Sorsa M, Einistö P. No difference between urinary mutagenicity in smokers of low-tar and medium-tar cigarettes: a double-blind cross-over study. Archives of Toxicology. 1986;(Suppl 9):115–9. [PubMed: 3468891]
-
Tuttle AM, Stämpfli M, Foster WG. Cigarette smoke causes follicle loss in mice ovaries at concentrations representative of human exposure. Human Reproduction. 2009;24(6):1452–9. [PubMed: 19228760]
-
10th Report on Carcinogens. US Department of Health and Human Services. Research Triangle Park (NC): U.S. Department of Health and Human Services, Public Health Service, National Toxicology Program; 2002.
-
US Department of Health and Human Services. The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2004.
-
U.S. Environmental Protection Agency. Ambient Water Quality Criteria for Polynuclear Aromatic Hydrocarbons. Washington: U.S. Environmental Protection Agency, Office of Water Regulations and Standards; 1980. Publication No. EPA 440/5-80-069.
-
U.S. Environmental Protection Agency. Quality Criteria for Water 1986. Washington: U.S. Environmental Protection Agency, Office of Water Regulations and Standards; 1986. Publication No. EPA 440/5-86-001.
-
van Doorn R, Leujdekkers CM, Bos RP, Brouns RME, Henderson PT. Detection of human exposure to electro-philic compounds by assay of thioether detoxification products in urine. Annals of Occupational Hygiene. 1981;24(1):77–92. [PubMed: 7015964]
-
van Jaarsveld H, Kuyl JM, Alberts DW. Exposure of rats to low concentration of cigarette smoke increases myocardial sensitivity to ischaemia/reperfusion. Basic Research in Cardiology. 1992;87(4):393–9. [PubMed: 1417708]
-
Van Rooij JGM, Veeger MMS, Bodelier-Bade MM, Scheepers PTJ, Jongeneelen FJ. Smoking and dietary intake of polcyclic aromatic hydrocarbons as sources of interindividual variability in the baseline excretion of 1-hydroxypyene in urine. International Archives of Occupational and Environmental Health. 1994;66(1):55–65. [PubMed: 7927844]
-
Van Schooten FJ, Hirvonen A, Maas LM, De Mol BA, Kleinjans JCS, Bell DA, Durrer JD. Putative susceptibility markers of coronary artery disease: association between VDR genotype, smoking, and aromatic DNA adduct levels in human right atrial tissue. FASEB Journal. 1998;12(13):1409–17. [PubMed: 9761785]
-
Wang H. Differential responses of Paramecium aurelia to cigarette smoke. Nature. 1963;197(4871):946–8. [PubMed: 13998781]
-
Wanner A. A review of the effects of cigarette smoke on airway mucosal function. European Journal of Respiratory Disease. 1985;66(Suppl 139):49–53. [PubMed: 3862611]
-
Ward E, Carpenter A, Markowitz S, Roberts D, Halperin W. Excess number of bladder cancers in workers exposed to ortho-toluidine and aniline. Journal of the National Cancer Institute. 1991;83(7):501–6. [PubMed: 2005633]
-
Watson ID. Rapid analysis of nicotine and cotinine in the urine of smokers by isocratic high-performance liquid chromatography. Journal of Chromatography B. 1977;143(2):203–6. [PubMed: 838832]
-
Watson CH, Trommel JS, Ashley DL. Solid-phase microextraction-based approach to determine free-base nicotine in trapped mainstream cigarette smoke total particulate matter. Journal of Agricultural and Food Chemistry. 2004;52(24):7240–5. [PubMed: 15563201]
-
Wayne GF, Connolly GN. Application, function, and effects of menthol in cigarettes: a survey of tobacco industry documents. Nicotine & Tobacco Research. 2004;6( Suppl 1):S43–S54. [PubMed: 14982708]
-
Wayne GF, Connolly GN, Henningfield JE, Farone WA. Tobacco industry research and efforts to manipulate particle size: implications for product regulation. Nicotine & Tobacco Research. 2008;10(4):613–25. [PubMed: 18418784]
-
Weiss W, Weiss WA. Effect of tobacco smoke solutions on Paramecium. Archives of Environmental Health. 1964;9:500–4. [PubMed: 14185556]
-
Wennmalm Å, Benthin G, Granström EF, Persson L, Petersson A-S, Winell S. Relation between tobacco use and urinary excretion of thromboxane A2 and prostacyclin metabolites in young men. Circulation. 1991;83(5):1698–704. [PubMed: 2022025]
-
Williamson JT, Graham JF, Allman DR. The modification of cigarette smoke by filter tips. Beiträge zur Tabakforschung International. 1965;3(3):233–42.
-
Wistuba II, Lam S, Behrens C, Virmani AK, Fong KM, LeRiche J, Samet JM, Srivastava S, Minna JD, Gazdar AF. Molecular damage in the bronchial epithelium of current and former smokers. Journal of the National Cancer Institute. 1997;89(18):1366–73. [PMC free article: PMC5193483] [PubMed: 9308707]
-
Witschi H. A/J mouse as a model for lung tumorigenesis caused by tobacco smoke: strengths and weaknesses. Experimental Lung Research. 2005;31(1):3–18. [PubMed: 15765916]
-
Witschi H, Espiritu I, Dance ST, Miller MS. A mouse lung tumor model of tobacco smoke carcinogenesis. Toxicological Sciences. 2002;68(2):322–30. [PubMed: 12151628]
-
Witschi H, Espiritu I, Maronpot RR, Pinkerton KE, Jones AD. The carcinogenic potential of the gas phase of environmental tobacco smoke. Carcinogenesis. 1997a;18(11):2035–42. [PubMed: 9395199]
-
Witschi H, Espiritu I, Peake JL, Wu K, Maronpot RR, Pinkerton KE. The carcinogenicity of environmental tobacco smoke. Carcinogenesis. 1997b;18(3):575–86. [PubMed: 9067559]
-
Witschi H, Espiritu I, Uyeminami D, Suffia M, Pinkerton KE. Lung tumor response in strain A mice exposed to tobacco smoke: some dose-effect relationships. Inhalation Toxicology. 2004;16(1):27–32. [PubMed: 14744662]
-
Woodman G, Newman SP, Pavia D, Clarke SW. Inhaled smoke volume, puffing indices and carbon monoxide uptake in asymptomatic cigarette smokers. Clinical Science (London). 1986;71(4):421–7. [PubMed: 3757438]
-
World Tobacco Staff. Tobacco's essential partner. World Tobacco. 2000 Mar;174:21–2.
-
Wu W, Zhang L, Jain RB, Ashley DL, Watson CH. Determination of carcinogenic tobacco-specific nitrosamines in mainstream smoke from U.S.-brand and non-U.S.-brand cigarettes from 14 countries. Nicotine & Tobacco Research. 2005;7(3):443–51. [PubMed: 16085512]
-
Wu X, Lippmann SM, Lee JJ, Zhu Y, Wei QV, Thomas M, Hong WK, Spitz MR. Chromosome instability in lymphocytes: a potential indicator of predisposition to oral premalignant lesions. Cancer Research. 2002;62(10):2813–8. [PubMed: 12019158]
-
Wynder EL, Goodman DA, Hoffmann D. Ciliatoxic components in cigarette smoke. 3: in vitro comparison of different smoke components. Cancer. 1965;18(12):1652–8. [PubMed: 5845804]
-
Wynder EL, Graham EA, Croninger AB. Experimental production of carcinoma with cigarette tar. Cancer Research. 1953;13(12):855–64. [PubMed: 13116124]
-
Wynder EL, Hoffmann D. A study of tobacco carcinogenesis. VIII: the role of the acidic fractions as promoters. Cancer. 1961;14(6):1306–15. [PubMed: 14008628]
-
Wynder EL, Hoffmann D. New York: Academic Press; Tobacco and Tobacco Smoke: Studies in Experimental Carcinogenesis. 1967
-
Wynder EL, Kopf P, Ziegler H. A study of tobacco carcinogenesis. II: dose-response studies. Cancer. 1957;10(6):1193–200. [PubMed: 13489670]
-
Xiao T, Guha J, Boyle D, Liu C-Q, Chen J. Environmental concerns related to high thallium levels in soils and thallium uptake by plants in southwest Guizhou, China. Science of the Total Environment. 2004a;318( 1–3):223–44. [PubMed: 14654287]
-
Xiao T, Guha J, Boyle D, Liu C-Q, Zheng B, Wilson GC, Rouleau A, Chen J. Naturally occurring thallium: a hidden geoenvironmental health hazard? Environment International. 2004b;30(4):501–7. [PubMed: 15031009]
-
Xu L, Cai B-Q, Zhu Y-J. Pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease and therapeutic effects of glucocorticoids and N-acet-ylcysteine in rats. Chinese Medical Journal. 2004;117(11):1611–9. [PubMed: 15569474]
-
Yamasaki E, Ames BN. Concentration of mutagens from urine by adsorption with the nonpolar resin XAD-2: cigarette smokers have mutagenic urine. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(8):3555–9. [PMC free article: PMC431630] [PubMed: 333441]
-
Yang IY, Chan G, Miller H, Huang Y, Torres MC, Johnson F, Moriya M. Mutagenesis by acrolein-derived propano-deoxyguanosine adducts in human cells. Biochemistry. 2002;41(46):13826–32. [PubMed: 12427046]
-
Yauk CL, Berndt ML, Williams A, Rowan-Carroll A, Douglas GR, Stampfli MR. Mainstream tobacco smoke causes paternal germ-line DNA mutations. Cancer Research. 2007;67(11):5103–6. [PubMed: 17545587]
-
Yoon J-H, Smith LE, Feng Z, Tang M-S, Lee C-S, Pfeifer GP. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Research. 2001;61(19):7110–7. [PubMed: 11585742]
-
Zacny JP, Stitzer ML. Human smoking patterns, The FTC Cigarette Test Method for Determining Tar, Nicotine, and Carbon Monoxide Yields of US Cigarettes. Bethesda (MD): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute; 1996. pp. 151–60. Report of the NCI Expert Committee Panel Smoking and Tobacco Control Monograph No 7. NIH Publication No 96-4028.
-
Zacny JP, Stitzer ML, Brown FJ, Yingling JE, Griffiths RR. Human cigarette smoking: effects of puff and inhalation parameters on smoke exposure. Journal of Pharmacology and Experimental Therapeutics. 1987;240(2):554–64. [PubMed: 3806411]
-
Zacny JP, Stitzer ML, Yingling JE. Cigarette filter vent blocking: effects on smoking topography and carbon monoxide exposure. Pharmacology, Biochemistry, and Behavior. 1986;25(6):1245–52. [PubMed: 3809227]
-
Zaliznyak T, Bonala R, Attaluri S, Johnson F, de los Santos C. Solution structure of DNA containing α-OH-PdG: the mutagenic adduct produced by acrolein. Nucleic Acids Research. 2009;37(7):2153–63. [PMC free article: PMC2673425] [PubMed: 19223332]
-
Zhang S, Villalta PW, Wang M, Hecht SS. Detection and quantitation of acrolein-derived 1,N 2 -propanodeoxy-guanosine adducts in human lung by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chemical Research in Toxicology. 2007;20(4):565–71. [PMC free article: PMC2518976] [PubMed: 17385896]
-
Zhu Y, Spitz MR, Zheng Y-L, Hong WK, Wu X. BPDE-induced lymphocytic 3p21.3 aberrations may predict head and neck carcinoma risk. Cancer. 2002;95(3):563–8. [PubMed: 12209748]
How to Get Rid of Black Lips From Smoking Weed
Source: https://www.ncbi.nlm.nih.gov/books/NBK53014/